
Abstract
Background:
Uncomplicated acute rhinosinusitis (ARS) is usually a self-limiting inflammatory condition often treated with antibiotics.
Aims:
To assess the safety and efficacy of fluticasone furoate nasal spray (FFNS) compared with placebo for symptomatic relief of uncomplicated ARS.
Methods:
A randomised, double-blind, placebo-controlled, parallel-group, multicentre, 2-week treatment study of FFNS 110μg once and twice daily was undertaken in adults/adolescents.
Results:
A statistically significant reduction was seen in the daily major symptoms score, a composite score of three individual symptoms (nasal congestion/stuffiness, sinus headache/pressure or facial pain/pressure, and postnasal drip on a 0–3 scale) by both FFNS doses compared with placebo (least square mean differences vs. placebo of −0.386 (p=0.008) and −0.357 (p=0.014) for once daily and twice daily FFNS, respectively). The differences in median times to symptom improvement were not statistically significant between each dose of FFNS (7 days) and placebo (8 days). There were no treatment differences in antibiotic use for possible fulminant bacterial rhinosinusitis (3% in each group). The safety profile of FFNS was similar to placebo.
Conclusions:
FFNS reduces symptoms of uncomplicated ARS compared with placebo and is well tolerated, providing support for withholding antibiotics in selected patients.
Similar content being viewed by others


Introduction
Acute rhinosinusitis (ARS) is a common reason for primary care visits and causes significant symptoms, often resulting in work/school absences. It is defined as a sudden onset of ≥2 symptoms, one of which is nasal blockage/congestion or nasal discharge (anterior or posterior). Other symptoms are facial pain/pressure and impairment/loss of smell.1 These symptoms usually have an acute onset and are present for <4 weeks.2 ARS is distinguished from the common cold by persistent sinus inflammation after the usual 10- day period of recovery from a common cold or worsening after an initial period of improvement. The European guideline1 advises treatment of ARS depending on severity: to start with symptomatic relief for mild ARS and to add intranasal corticosteroids for moderate ARS. Antibiotics are added only when symptoms such as fever >38°C or severe pain are present.1
It has been theorised that an intranasal corticosteroid would promote drainage and increase aeration of the sinuses by decreasing the inflammatory response and reducing mucosal swelling, thus hastening the elimination of infectious organisms and decreasing the frequency/severity of recurrent symptoms.3 Several studies conclude that intranasal corticosteroids (with or without antibiotics) are beneficial in ARS patients and are equally or more effective than antibiotics alone.4–8 Currently, only one intranasal corticosteroid — mometasone furoate nasal spray (MFNS) — is approved for ARS,9 and this indication is limited to Canada. Fluticasone furoate, an enhanced-affinity glucocorticoid, has been developed as an intranasal spray for the treatment of allergic rhinitis (AR). The safety of fluticasone furoate nasal spray (FFNS) has been demonstrated in a 12-month study in adults/adolescents with perennial AR.10
The objective of this study was to evaluate the safety and efficacy of two doses of FFNS (110μg once daily and 110μg twice daily) compared with placebo as monotherapy in treating adults/adolescents with uncomplicated ARS. For this study, uncomplicated ARS was defined as persistent inflammation of the paranasal sinuses and nasal cavity beyond 10 days. The defined study population excluded subjects with fulminant bacterial rhinosinusitis (FBRS) clinically suggested by symptoms including temperature >38°C and persistent severe facial/tooth pain. Subjects with symptomatic AR and other sinonasal conditions including chronic or recurrent rhinosinusitis were also excluded to ensure only those with ARS were studied.
Methods
Study design
This randomised, double-blind, placebo-controlled, parallel-group study was carried out at 67 sites in 12 countries (Table 1). It was conducted according to the International Conference on Harmonisation, Good Clinical Practice and all applicable subject privacy requirements, and the ethical principles as outlined in the Declaration of Helsinki 2008. For the completed CONSORT checklist, see Appendix 1 available online at www.thepcrj.org
Males or non-pregnant females aged ≥12 years with uncomplicated ARS were eligible. Subjects could not have a clinical diagnosis of FBRS, other concurrent sinonasal conditions including chronic or recurrent rhinosinusitis, or symptomatic AR or allergic sensitisation to seasonal allergens likely to be present during the study (determined by skin prick test or in vitro blood test).
Subjects completed a diary (see Appendix 2, available online at www.thepcrj.org) in which they rated the symptom severity of ARS based on the major symptom score (MSS), a composite score of three individual symptoms (nasal congestion/stuffiness, sinus headache/pressure or facial pain/pressure, postnasal drip), each using a scale of 0–3. These assessments were conducted twice daily, before the morning and evening dose, approximately 12 hrs apart. Subjects also documented compliance with study drug, medical conditions, and concomitant medications taken during the study.
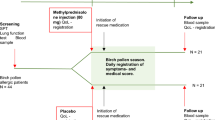
Five clinic visits were scheduled (Figure 1). Subjects who were experiencing ARS symptoms for 5–8 days before Visit 1 and met the inclusion criteria entered a 3–5 day screening period. At the end of the screening period, subjects who had an average of ≥4.5 on the MSS and ≥2 on congestion/stuffiness and sinus headache/pressure or facial pain/pressure (the last six assessments) were eligible for randomisation.

Study schematic. BD=twice daily, OD=once daily.
Eligible subjects were randomly assigned (1:1:1) to receive one of three double-blinded (to subjects/care givers, investigators, and sponsor study personnel) study treatments (FFNS 110μg once daily, FFNS 110μg twice daily, or placebo) for 2 weeks, according to a computer-generated randomisation schedule and an Interactive Voice Response System called the Registration and Medication Ordering System. The randomisation was stratified by country, age (<18 years, ≥18 years), and AR status (yes/no). Subjects were not permitted to take any medications that may affect the duration/severity of rhinosinusitis throughout the screening/treatment period. The subjects attended the clinic for Visits 3 (1 week on treatment), 4 (end of 2 weeks of treatment), and 5 (1 week post-treatment follow-up). Subjects received a follow-up telephone contact 7 days after Visit 5 for assessment of adverse events (AEs).
Assessments
The primary efficacy endpoint was the mean change from baseline in the daily MSS over the entire treatment period (weeks 1–2).
The key secondary endpoint was the first time to symptom improvement (defined as reduction of individual symptom scores of nasal congestion/stuffiness, sinus headache/pressure or facial pain/pressure, and postnasal drip to ≤1 for two consecutive 12-hr assessments). The mean change from baseline over the entire treatment period in morning and evening MSS and individual symptom scores was also evaluated as secondary endpoints. The use of an antibiotic during the study for FBRS was also evaluated. Exploratory endpoints included mean change from baseline over the entire treatment period in daily, morning and evening individual symptom scores for purulent rhinorrhoea and cough.
Safety assessments included AEs, nasal examination (the mucosa for bleeding, ulcers, polyposis, or candidiasis), clinical laboratory tests, and vital signs (blood pressure, heart rate, and temperature).
Health outcome endpoints included mean change from baseline in total Sino-Nasal Outcomes Test-20 (SNOT-20)11 score (range 0–5) at the end of treatment and an assessment of changes in productivity and sleep using a daily diary over the entire treatment period on a scale of 0–10. (See Appendix 2, available online at www.thepcrj.org)
Statistical methods
The proposed sample size of 240 subjects per treatment group was estimated to provide 90% power to detect a difference of 0.45 between FFNS (either dose) and placebo in mean change from baseline over the entire treatment period in daily MSS at a two-sided significance level of 0.05 assuming a standard deviation of 1.525.
All analyses and summaries were based on the intent-to-treat population (ITT; randomised and received at least one dose of study medication). The primary analysis method was a pairwise comparison between each FFNS dose and placebo using analysis of covariance with adjustments for baseline value, country, AR status, age, and gender. Time to symptom improvement was analysed using Kaplan-Meier estimates and the pairwise comparisons between each active group and placebo was performed based on a log-rank test.
Results
Subject disposition and baseline characteristics
The study was conducted from January to July 2010. Recruitment ended soon after the study reached the enrolment goal (720 subjects). A total of 1023 subjects were screened, of which 737 subjects were randomised and received one of three treatments. Baseline characteristics and symptom severity were similar among the three treatment groups (Tables 1 and 2). Thirty-nine subjects prematurely withdrew from study treatment; 95% of the 737 subjects in the analyses completed the 2-week study treatment. The most common reason for withdrawal was AEs (4% in placebo group, 2% in FFNS 110μg once daily group, and 2% in FFNS 110μg twice daily group) (Figure 2).

Study population. BD=twice daily, OD=once daily
Efficacy
For the primary efficacy endpoint, the mean daily MSS declined (improved) in all three treatment groups during the 2-week treatment period (Figure 3). A statistically significant reduction in daily MSS by both FFNS doses was seen compared with placebo (least squares mean differences vs. placebo of −0.386 (p=0.008) and −0.357 (p=0.014) for once daily and twice daily FFNS 110μg, respectively). Treatment differences in morning and evening MSS were also significant for both FFNS doses compared with placebo (Table 2).

Least squares mean (SE) change from baseline in daily major symptom score (MSS) during the 2-week treatment period. BD=twice daily, OD=once daily, FFNS, fluticasone furoate nasal spray
Over the course of treatment, reductions from baseline in daily symptom scores for all three major individual symptoms were observed in all treatment groups. Treatment differences compared with placebo were significant for both FFNS doses for the daily nasal congestion/stuffiness score and only for once daily FFNS 110μg for the daily postnasal drip score. No treatment differences were observed in the daily sinus headache/pressure or facial pain/pressure score. Treatment differences in morning and evening individual symptom scores were similar to those for daily individual symptom scores.
For the key secondary endpoint, the median time to symptom improvement was 8 days for the placebo group and 7 days for each of the FFNS groups. The treatment differences between the two doses of FFNS and placebo were not statistically significant.
There were no treatment differences in antibiotic use for FBRS among the three treatment groups (seven subjects (3%) in each treatment group; Table 3).
Safety
The incidence of AEs during the treatment period was similar across all treatment groups with the most common AEs being headache, bacterial sinusitis, and epistaxis (Table 4). There were no deaths. One serious AE was reported which was not drug-related. Epistaxis was the most common drug-related AE (2% in placebo group, 3% in FFNS 110μg once daily group, and <1% in FFNS 110μg twice daily group during treatment). Headache was also reported as a drug-related AE (<1% in placebo group, 1% in FFNS 110μg once daily group, and 2% in FFNS 110μg twice daily group during treatment.
More subjects in the placebo group (7%) prematurely withdrew from study treatment than in the FFNS groups (3% and 5% for once daily and twice daily treatment groups, respectively; Figure 2). Twenty subjects were withdrawn from study treatment due to AEs, with the most common one being bacterial sinusitis (2% in the placebo group, <1% in the FFNS 110μg once daily group, and 1% in the FFNS 110μg twice daily group).
There were no significant findings from clinical laboratory tests and vital signs. Most nasal examinations showed no change from baseline to the end of study treatment/early withdrawal. Mucosal bleeding was the most common abnormal finding at endpoint (worsening in 2% of subjects in each treatment group). Four subjects had evidence of possible nasal candidiasis during the study (1% in the placebo group and 1% in the FFNS 110μg twice daily group).
Health outcomes
Mean changes from baseline in total SNOT-20 scores (Table 5) at the end of treatment did not show statistically significant differences between either FFNS dosage group and the placebo group (least squares mean difference vs. placebo −0.110 (p=0.142) and −0.142 (p=0.058) for FFNS 110μg once daily and twice daily, respectively). The mean changes from baseline over weeks 1–2 were statistically significant between both FFNS doses and placebo for productivity (least squares mean difference vs. placebo for FFNS 110μg once daily and twice daily −0.293 (p=0.049) and −0.385 (p=0.010), respectively) and sleep (-0.324 (p=0.038) and −0.343 (p=0.027), respectively), based on daily diary.
Discussion
Although uncomplicated ARS is usually a self-limiting inflammatory condition, it is often treated with antibiotics.12 Treatment recommendations for ARS vary from only treating severe/persistent moderate symptoms and specific bacterial rhinosinusitis findings with narrow spectrum antibiotics to treating all patients with broad spectrum antibiotics.1,13 This study evaluated an alternative treatment for symptomatic relief of uncomplicated ARS using an intranasal corticosteroid, fluticasone furoate (FFNS), as monotherapy. The advantages of this therapeutic approach are two-fold: eliminating unnecessary antibiotic use that aggravates bacterial resistance: and providing symptom control in patients for whom antibiotics have been shown to provide limited benefit.14 This hypothesis was supported by previous controlled trials that have shown the benefit of intranasal corticosteroids with their recognised anti-inflammatory properties, with or without antibiotics in ARS.15
Main findings
This study demonstrated a statistically significant treatment benefit of both FFNS once daily and twice daily doses compared with placebo in reducing the overall symptoms of uncomplicated ARS based on the primary efficacy endpoint, the mean change from baseline over the entire treatment period in daily MSS.
Among the study population, as expected for a self-limiting condition, all treatment groups showed a reduction in symptom severity during the treatment period (within 4 weeks from onset). The lack of statistically significant differences in time to symptom improvement between each FFNS dose and placebo (1 day difference) may be attributable to the selection of study subjects in addition to the self-limiting nature of the condition. In this population with ARS who did not have chronic or recurrent rhinosinusitis, it could have been more difficult to show a difference between FFNS and placebo by the rate of symptom improvement than if it was examined in a pool of subjects with pre-existing symptoms less likely to resolve spontaneously. Likewise, exclusion of chronic or recurrent rhinosinusitis and symptomatic AR eliminated subjects who may have had more severe symptoms and, therefore, could have had more pronounced benefit from the anti-inflammatory action of an intranasal corticosteroid. Among the study population without co-morbid sinonasal conditions, the clinical effect of FFNS in reducing symptoms compared with placebo was modest and not supported by a more rapid improvement in symptoms.
In evaluating the impact on quality of life using SNOT-20 (a questionnaire validated in chronic rhinosinusitis), there was no treatment benefit of FFNS based on the total SNOT-20 score (1-week recall). However, based on daily diary questions, FFNS showed treatment benefit compared with placebo in productivity and sleep.
Both FFNS doses were well tolerated based on safety assessments. AE findings from the study were similar to those from other short-term treatment studies using FFNS in subjects with AR.
Interpretation of findings in relation to previously published work
Compared with other studies using the intranasal corticosteroid MFNS (the MFNS study8,9), this FFNS study differs in several key inclusion/exclusion criteria (Table 6). In this FFNS study, most subjects (98%) had symptoms for 8–13 days before entering study treatment whereas, in the MFNS study, subjects could have symptoms for up to 28 days and 23–28% in each group had symptoms for 15–28 days before starting study treatment. The FFNS study explicitly excluded subjects with symptomatic perennial AR and seasonal AR as well as those sensitised to seasonal allergens that could be present during the study. In contrast, it is not clear whether the MFNS study excluded subjects with symptomatic perennial AR. In addition, the MFNS study did not specifically exclude subjects with a history of recurrent rhinosinusitis whereas the FFNS study excluded subjects with current or a history of sinonasal conditions including chronic or recurrent rhinosinusitis. The MFNS studies suggested that the twice daily dose was efficacious while the once daily dose was insufficient to reduce the ARS symptoms.8,9 In another study, budesonide — which is less topically potent than FFNS or MFNS — given once daily had no benefit over placebo for ARS.16 The differences in efficacy between these studies, including the relative potency of once daily compared with twice daily dosing, may be due to the differences in study population and the potency of the individual intranasal corticosteroid as well as symptoms comprising a MSS. Despite the carefully selected uncomplicated ARS population that would be most likely to improve spontaneously, this study demonstrated statistically significant treatment benefit of both FFNS once daily and twice daily regimens compared with placebo.
The subject selection criteria, reflecting a clinical diagnosis of uncomplicated ARS and excluding FBRS without imaging techniques or sinus aspirate culture, was able to identify a pool of patients with uncomplicated ARS who did not require antibiotics for symptom relief. In the FFNS study the majority of subjects with uncomplicated ARS (97% in each group) did not require an antibiotic for FBRS. The previous MFNS study in ARS subjects comparing an antibiotic and intranasal corticosteroid treatments to placebo had similar results (treatment failure rate for MFNS twice daily 4.7% compared with 7.2% for amoxicillin (p=0.258).8 This result suggests that administration of FFNS in subjects with uncomplicated ARS did not increase the risk of developing symptoms that may require antibiotic therapy. No significant difference in premature withdrawal from study treatment due to bacterial sinusitis between the FFNS and placebo groups also supports the safety of FFNS.
Strengths and limitations of this study
To our knowledge, this is the first large-scale, randomised, placebo-controlled study of uncomplicated ARS where patients with other co-morbid sinonasal conditions — including chronic or recurrent rhinosinusitis and symptomatic AR — have clearly been excluded. Because the clinical symptoms of these conditions overlap, a carefully selected patient population was critical to ensure that an observed treatment effect could be considered an effect on the ARS symptoms rather than on other pre-existing conditions.
The difficulties encountered included finding subjects with AR who were not exposed to relevant allergens during the study, and those who had appropriate ARS symptoms for a sufficient time to exclude acute viral illnesses and were willing to avoid other treatments for their condition. It was felt necessary to avoid the effects of any other treatments as the placebo nasal spray would already have a benefit when given twice daily. However, such selection criteria could have increased inclusion of subjects with milder symptoms who could tolerate symptoms without concurrent therapy. Alternative methodologies that would have been helpful to address the research question include allowing other medications for the condition and comparison with any additional benefit provided by intranasal corticosteroids, although the self-limiting nature of ARS would have made it more difficult to demonstrate treatment differences compared with placebo on top of other symptom relief medications. Allowing subjects with symptomatic perennial AR would have made recruitment much easier, but it would have made it difficult to address the question of the efficacy of FFNS in uncomplicated ARS separately from its proven effectiveness in relieving symptoms of perennial AR. Subjects with recurrent sinusitis would be another group worth studying since there are limited treatment options for these patients.
Implications for future research and practice
New questions arising from this study include: the optimal dose of FFNS needed to relieve uncomplicated ARS symptoms, depending on the symptom severity at the time of initial diagnosis, especially among patient groups that may not have been eligible for the study; whether patients with persistent ARS symptoms for a longer period, possibly due to their sinonasal co-morbidity, can benefit from FFNS 110μg twice daily or even a higher dose; and whether mild symptoms can be addressed by FFNS 55μg once daily. A validated health outcomes questionnaire in the study population is also needed to understand better the impact of FFNS treatment for ARS on quality of life. Lessons for clinical practice from this study include the benefits of using a potent intranasal corticosteroid such as FFNS which has a long duration of effect when give once daily,17 a similar benefit to that seen in treating the symptoms of AR, while avoiding antibiotics.
Conclusions
This study has demonstrated the efficacy of an FFNS given once or twice daily as an effective monotherapy in uncomplicated ARS. The study population was reflective of patients with uncomplicated ARS who can be identified based on the clinical diagnosis in a primary care setting. In this population, both FFNS doses demonstrated a statistically significant treatment difference compared with placebo in reducing overall symptoms of uncomplicated ARS. Furthermore, the safety profile of FFNS in uncomplicated ARS was similar to that in AR. In addition, the low incidence of infections requiring antibiotics provided valuable clinical support for emerging treatment guidelines that recommend withholding antimicrobial treatment for patients with uncomplicated ARS.
References
Fokkens W, Lund V, Mullol J, on behalf of the European Position Paper on Rhinosinusitis and Nasal Polyps Group. European position paper on rhinosinusitis and nasal polyps 2007. Rhinology 2007;(Suppl 20):1–136.
Rosenfeld RM, Andes D, Bhattacharyya N, et al. Clinical practice guideline: adult sinusitis. Otolaryngol Head Neck Surg 2007;137(3 Suppl):S1–31. http://dx.doi.org/10.1016/j.otohns.2007.06.726
Mygind N, Prytz S, Sorensen H, Pedersen CB . Long-term treatment of nasal polyps with beclomethasone dipropionate aerosol. I. Treatment and rationale. Acta Otolaryngol 1976;82(3–4):252–5. http://dx.doi.org/10.3109/00016487609120897
Barlan IB, Erkan E, Bakir M, Berrak S, Basaran MM . Intranasal budesonide spray as an adjunct to oral antibiotic therapy for acute sinusitis in children. Ann Allergy Asthma Immunol 1997;78(6):598–601. http://dx.doi.org/10.1016/S1081-1206(10)63223-1
Dolor RJ, Witsell DL, Hellkamp AS, Williams JW Jr, Califf RM, Simel DL, Ceftin and Flonase for Sinusitis (CAFFS) Investigators. Comparison of cefuroxime with or without intranasal fluticasone for the treatment of rhinosinusitis. The CAFFS Trial: a randomized controlled trial. JAMA 2001;286(24):3097–105. http://dx.doi.org/10.1001/jama.286.24.3097
Meltzer EO, Orgel HA, Backhaus JW, et al. Intranasal flunisolide spray as an adjunct to oral antibiotic therapy for sinusitis. J Allergy Clin Immunol 1993;92(6):812–23. http://dx.doi.org/10.1016/0091-6749(93)90058-N
Meltzer EO, Charous BL, Busse WW, Zinreich SJ, Lorber RR, Danzig MR . Added relief in the treatment of acute recurrent sinusitis with adjunctive mometasone furoate nasal spray. The Nasonex Sinusitis Group. J Allergy Clin Immunol 2000;106(4):630–7. http://dx.doi.org/10.1067/mai.2000.109056
Meltzer EO, Bachert C, Staudinger H . Treating acute rhinosinusitis: comparing efficacy and safety of mometasone furoate nasal spray, amoxicillin, and placebo. J Allergy Clin Immunol 2005;116(6):1289–95. http://dx.doi.org/10.1016/j.jaci.2005.08.044
Merck Canada Inc. Nasonex mometasone furoate monohydrate aqueous nasal spray product monograph. 4 February 2011. Search product, Nasonex, on Health Canada Drug Product Database: http://webprod.hc-sc.gc.ca/dpd-bdpp/newSearch-nouvelleRecherche.do?lang=eng
Rosenblut A, Bardin PG, Muller B, et al. Long-term safety of fluticasone furoate nasal spray in adults and adolescents with perennial allergic rhinitis. Allergy 2007;62(9):1071–7. http://dx.doi.org/10.1111/j.1398-9995.2007.01521.x
Piccirillo JF, Merritt MG Jr, Richards ML . Psychometric and clinimetric validity of the 20-item Sino-Nasal Outcome Test (SNOT-20). Otolaryngol Head Neck Surg 2002;126(1):41–7. http://dx.doi.org/10.1067/mhn.2002.121022
Ryan D . Management of acute rhinosinusitis in primary care: changing paradigms and the emerging role of intranasal corticosteroids. Prim Care Respir J 2008;17(3):148–55. http://dx.doi.org/10.3132/pcrj.2008.00050
Snow V, Mottur-Pilson C, Hickner JM, American Academy of Family Physicians, American College of Physicians-American Society of Internal Medicine, Centers for Disease Control, Infectious Diseases Society of America. Principles of appropriate antibiotic use for acute sinusitis in adults. Ann Intern Med 2001;134(6):495–7.
Ahovuo-Saloranta A, Borisenko OV, Kovanen N, et al. Antibiotics for acute maxillary sinusitis. Cochrane Database Syst Rev 2008;(2):CD000243.
Zalmanovici A, Yaphe J . Steroids for acute sinusitis. Cochrane Database Syst Rev 2007;(2):CD005149.
Williamson IG, Rumsby K, Benge S, et al. Antibiotics and topical nasal steroid for treatment of acute maxillary sinusitis: a randomized controlled trial. JAMA 2007;298(21):2487–96. http://dx.doi.org/10.1001/jama.298.21.2487
Salter M, Biggadike K, Mathews JL, et al. Pharmacological properties of the enhanced-affinity glucocorticoid fluticasone furoate in vitro and in an in vivo model of respiratory inflammatory disease. Am J Physiol Lung Cell Mol Physiol 2007;293(3):L660–7. http://dx.doi.org/10.1152/ajplung.00108.2007
Acknowledgements
Handling editor Arnulf Langhammer
Statistical review Gopal Netuveli
The authors would like to thank Drs Edward Philpot and Bertrand Sohier and the FFS113203 study team for their contribution to the study.
Funding This study (FFS113203, NCT01018030) was funded by GlaxoSmithKline.
Author information
Authors and Affiliations
Contributions
All authors were involved in the initial concept and writing of the paper and retained full editorial control throughout the development of the manuscript. Final approval was endorsed by the authors. All authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors.
Corresponding author
Ethics declarations
Competing interests
PKK has received rhinitis-related research funding from GlaxoSmithKline, Merck, Nycomed, Allergy Therapeutics and Allergopharma. He has served on an advisory board for GlaxoSmithKline, Merck, Nycomed and received speakers' honoraria from GlaxoSmithKline, Merck, and Nycomed for rhinitis-related talks. AD has received research funding from Altana/Nycomed, Astra Zeneca, Bohringer Ingelheim, Chesi, Encorium, Fujisawa, GlaxoSmithKline, Hexal, LEK, Mudipharma, Pfizer, and UCB. OP has received research grants from ALK-Abello, Denmark; Allergopharma, Germany; Stallergenes, France; HAL, The Netherlands; Artu Biologicals, The Netherlands; Allergy-Therapeutics/Bencard, UK/Germany; Hartington, Spain; Lofarma, Italy; Novartis/Leti, Germany/Spain; GlaxoSmithKline, UK/Germany; Essex-Pharma, Germany; Cytos, Switzerland; Curalogic, Denmark; Roxall, Germany. He has also served as advisor and on the speakers' bureaus for some of the abovementioned pharmaceutical companies. WF has received research grants from GlaxoSmithKline, Stallargens, Medtronic, HAL, and Optinose. She has served on advisory boards for GlaxoSmithKline, MSD, and Stallargens and on speakers bureaus for GlaxoSmithKline, MSD, Stallargens, and Medtronics. SYK, WW, NT, CG, and LAL are full-time employees of GlaxoSmithKline and own stock in the company.
Appendices
Appendix 1: CONSORT 2010 checklist of information to include when reporting a randomised trial



Appendix 2: Diary cards












Rights and permissions
About this article
Cite this article
Keith, P., Dymek, A., Pfaar, O. et al. Fluticasone furoate nasal spray reduces symptoms of uncomplicated acute rhinosinusitis: a randomised placebo-controlled study. Prim Care Respir J 21, 267–275 (2012). https://doi.org/10.4104/pcrj.2012.00039
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.4104/pcrj.2012.00039
This article is cited by
-
Update on Intranasal Medications in Rhinosinusitis
Current Allergy and Asthma Reports (2017)
-
Diagnosis and Management of Rhinosinusitis: Highlights from the 2015 Practice Parameter
Current Allergy and Asthma Reports (2016)
