Targeting IgG in Arthritis: Disease Pathways and Therapeutic Avenues


1
School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510000, China
2
Department of Medical Biochemistry and Biophysics, Karolinska Institute, 17177 Stockholm, Sweden
Int. J. Mol. Sci. 2018, 19(3), 677; https://doi.org/10.3390/ijms19030677
Submission received: 31 December 2017
/
Revised: 25 January 2018
/
Accepted: 22 February 2018
/
Published: 28 February 2018
(This article belongs to the Special Issue Research of Pathogenesis and Novel Therapeutics in Arthritis)
Abstract
:Rheumatoid arthritis (RA) is a polygenic and multifactorial syndrome. Many complex immunological and genetic interactions are involved in the final outcome of the clinical disease. Autoantibodies (rheumatoid factors, anti-citrullinated peptide/protein antibodies) are present in RA patients’ sera for a long time before the onset of clinical disease. Prior to arthritis onset, in the autoantibody response, epitope spreading, avidity maturation, and changes towards a pro-inflammatory Fc glycosylation phenotype occurs. Genetic association of epitope specific autoantibody responses and the induction of inflammation dependent and independent changes in the cartilage by pathogenic autoantibodies emphasize the crucial contribution of antibody-initiated inflammation in RA development. Targeting IgG by glyco-engineering, bacterial enzymes to specifically cleave IgG/alter N-linked Fc-glycans at Asn 297 or blocking the downstream effector pathways offers new avenues to develop novel therapeutics for arthritis treatment.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
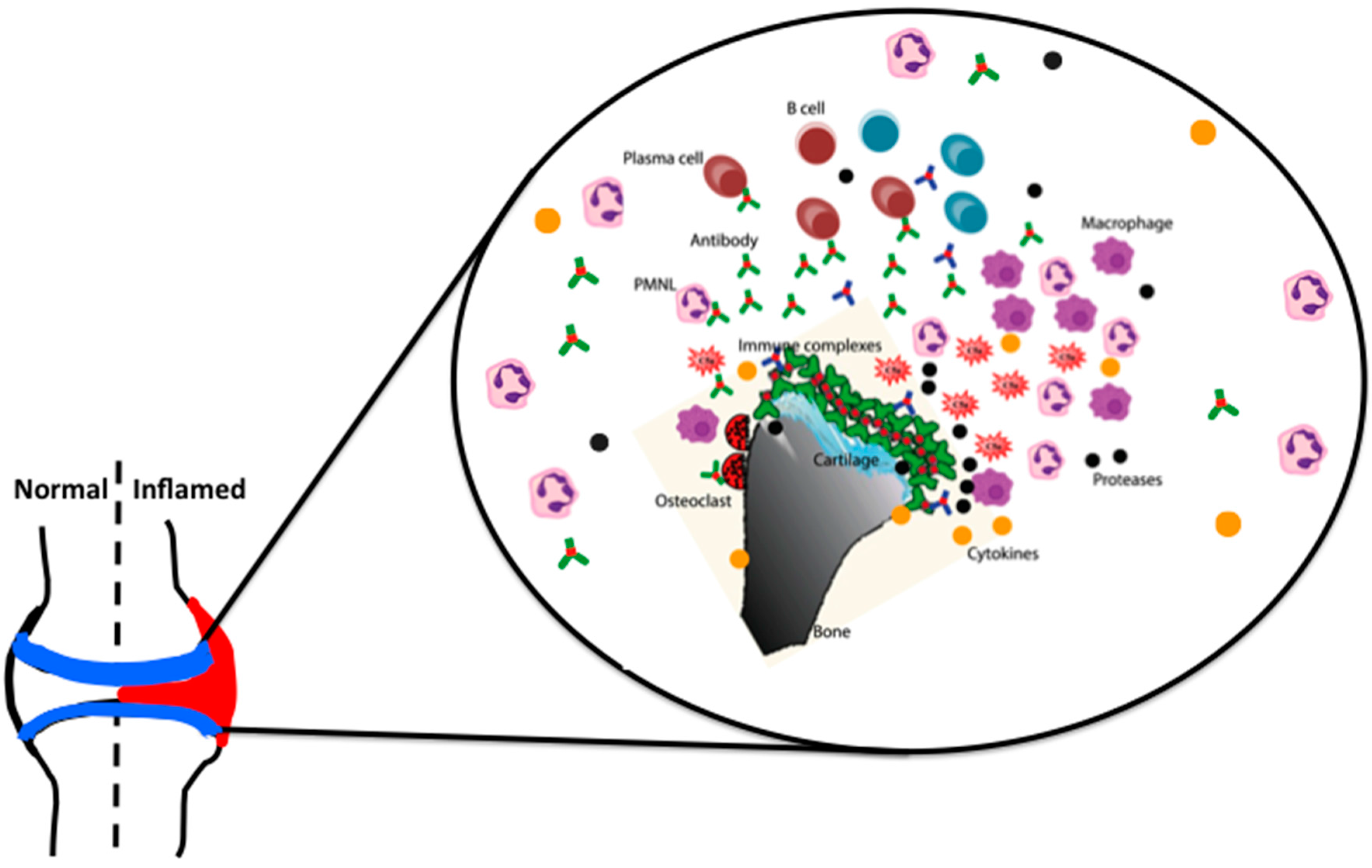
Rheumatoid arthritis (RA) in the articular joints involves a multicellular inflammatory process; infiltration of lymphocytes and granulocytes into the articular cartilage, proliferation of synovial cells, leukocyte extravasation, and, neo-vascularization of the synovial lining surrounding the joints [1]. This proliferative process not only induces swelling, erythema, and pain of multiple joints, but also progresses to the destruction and loss of cartilage and bone architecture. Many cellular components (macrophages, dendritic cells, synovial cells, mast cells, neutrophils, T cells, and B cells), cell surface molecules (co-receptors, adhesion molecules, and integrins), signaling components (ZAP70, PTPN22, JAK, MAPK and Stat1), metabolic components, and humoral mediators (antibodies, cytokines, chemokines, metalloproteinases, serine proteases, and aggrecanases) interact and aid in the disease progression, leading to the digestion of extracellular matrix and the destruction of articular structures [2].
Several theories on the pathogenesis of RA have been put forward that are based on autoantibodies and immune complexes, T cell mediated antigen specific immune responses, cytokine deregulations, and aggressive tumor-like behavior of the rheumatoid synovia. Improved understanding of the cellular and molecular events occurring in the rheumatoid joints during the pathogenesis of the disease is particularly important to find new or better combination of therapeutics for RA [3].
The major genetic factor that is consistently associated with RA is human leukocyte antigens (HLA), located on chromosome 6 in the major histocompatibility complex (MHC) class II region, which participate in the antigen presentation. DR genes, including DR4 and DR1 are associated with RA. The susceptibility epitope is glutamine-leucine-arginine-alanine-alanine (QKRAA) or glutamine-arginine-arginine-alanine-alanine (QRRAA), the so-called shared epitope identified in amino acids 70 through 74 in the third hypervariable region of the DRβ chain [2]. In addition, Raychaudhuri et al. have identified the amino acids (leucine or valine variants at amino acid position 11) that are located in the base of the antigen binding groove as further possible explanation for antigen selection [4]. The predominance of HLA and prominent infiltration of T cells to the rheumatoid synovia have suggested a key role for T cells in RA. Specific peptides that bind to these DR proteins in RA patients may promote arthritis, however, so far no such dominant peptides have been identified. It is possible that the susceptibility epitope is closely linked to other genes in the MHC region, and, T cells might drive the inflammation by their cellular interactions and cytokine production [5].
On the other hand, B cells contribute to the disease pathogenesis as antigen presenting cells, through co-stimulatory functions by supporting neo-lymphogenesis as well through the secretion of antibodies [6]. In RA, autoantibodies (rheumatoid factors (RFs), anti-citrullinated protein/peptide antibodies (ACPAs)) provide diagnostic and prognostic criteria, and serve as surrogate markers for disease activity), and may play a requisite role in the disease pathogenesis (anti-CII and anti-GPI antibodies) as well. RFs have been consistently associated with RA (60–80% sero-positivity), but it has also been reported to be present in normal individuals as well as under other chronic inflammatory conditions [7]. The contributions of antibodies to the disease are not solely dependent upon their direct binding to their respective antigens, but also through indirect mechanisms, including immune complex formation, deposition, and activation of complement components and FcγRs. Modulation of circulating ICs and pathogenic antibodies by removal using therapeutic plasmapheresis [8] or depleting B cells with the antibody rituximab proved to be beneficial for RA patients [9].
Most likely candidate autoantigens in RA are the joint derived macromolecules. Arthritis can be induced in animals by immunization with different components of cartilage; collagen type II (CII), collagen type IX (CIX), and collagen type XI (CXI), proteoglycan (PG), cartilage link protein (CLP), and chitinase 3-like protein 2 (CHI3L2/YKL-39). CII, a homo-trimer composed of α1(II) chains, is the most abundant fibrillar protein that is found in the articular cartilage and constitutes 80–85% of the total collagen. Autoimmunity to CII occurs in RA, target of inflammatory attack and CII has been proposed to be the driving force in arthritis [10].
Immunization of susceptible rodents with CII emulsified in adjuvant induced polyarthritis (so called, collagen induced arthritis, CIA), which resembles RA in several aspects. It has been well documented that both T and B cells are important in the disease pathogenesis, as demonstrated by the resistance of mice for arthritis induction that are deficient in these cell populations [11,12]. Similar to RA, susceptibility to CIA in rodents is closely associated with the expression of specific class II molecules of the MHC that are involved in the specific recognition of T cell receptor (TCR) and in binding and presenting antigenic peptides to it. Mice having H2q and H2r haplotypes are the most susceptible to arthritis [13]. Various humanized HLA transgenic mice having HLA-DQ8 [14], DR1 [15], or DR4 and CD4 [16] developed severe arthritis after CII immunization. In the H-2q context, the dominant heterologous T cell epitope resides in the amino acids position 260–270 [17,18]. Substitution of amino acids at positions 260-264 and 266 appeared to be critical for T cell recognition [19,20]. Interestingly, epitope glycosylation is important for T cell recognition of CII in CIA [21,22].
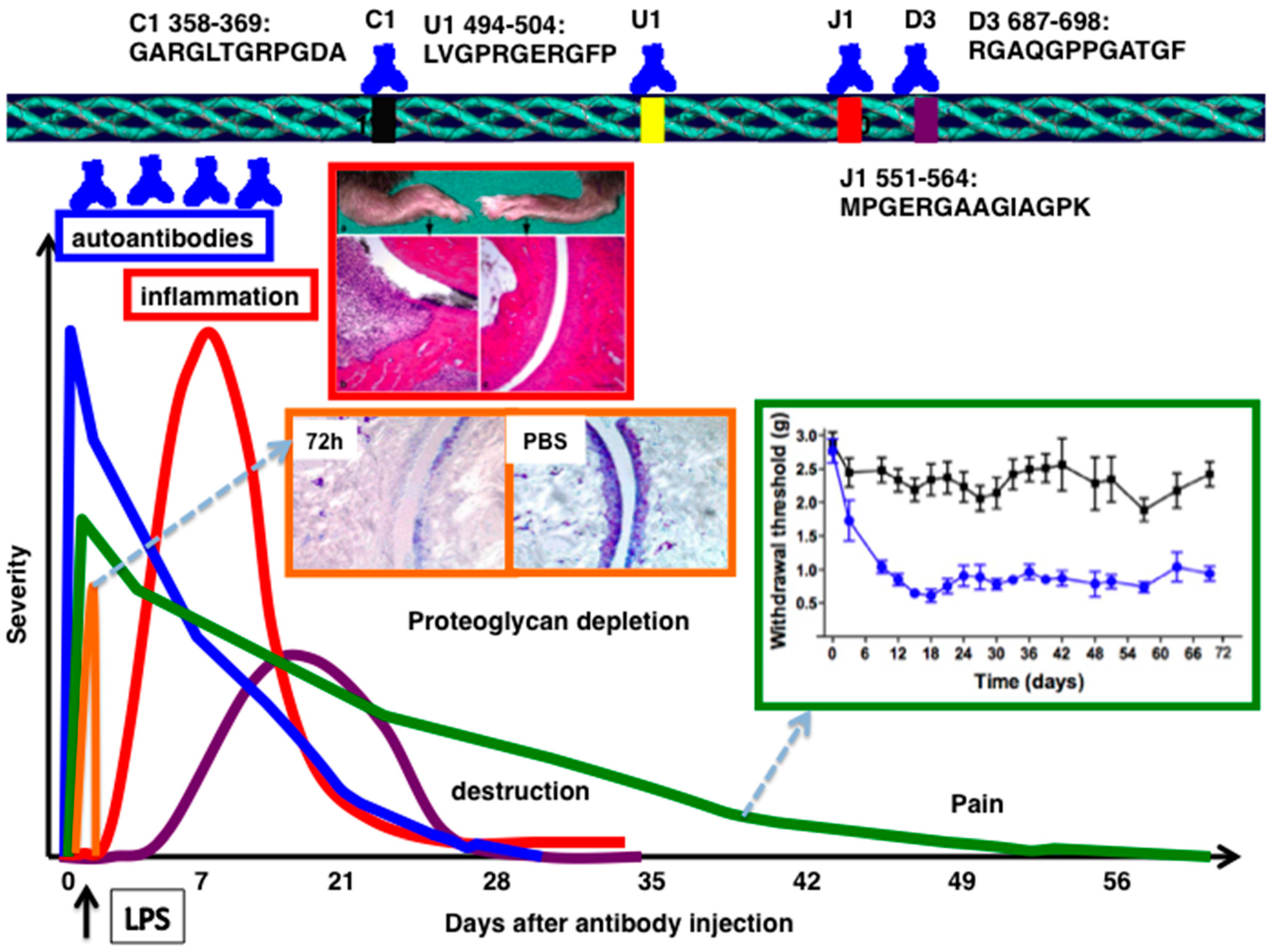
On the other hand, major B cell epitopes well defined so far (C1, J1, U1, D3, F4, and E8) are spread over the entire triple helical CII molecule. CII reactive B cells were shown to be neither negatively selected, somatically mutated, nor tolerized [23,24]. Native but not the denatured CII induces arthritis suggests the requirement of triple helical confirmation of CII for disease induction [25,26]. In CIA, antibodies play a major role in the immuno-pathology of autoimmune arthritis, and IgG and C3 depositions were detected in the inflamed joints [27,28]. Antibodies against C1, J1, and U1 epitopes were detected in CII immunized chronic arthritis mice [29], and these CII epitopes are conserved across the species [30]. However, DBA/1 mice deficient in the RAG1 gene still developed some synovial hyperplasia, pannus, and erosions of cartilage and bone [31], demonstrating that arthritis development is still possible even in the absence of mature T and B lymphocytes.
2. CII-Specific Antibodies
Germ line encoded antibodies are important in the pathogenesis of antibody mediated autoimmune diseases [32]. Genetic control of autoantibody responses [33,34] and the association of epitope-specific antibody response with specific VH alleles were identified earlier [35]. Antibodies either directly or as constituents of immune complexes, play a central role in triggering inflammation in a number of autoimmune diseases [6,36]. In experimental arthritis, disease can be passively induced in naive mice using serum from arthritic mice [27,37], RA patients [38,39], with a combination of CII specific mAbs [40,41,42,43,44] or single mAb [45]. Arthritis produced by passive transfer of CII mAb, so called collagen antibody induced arthritis (CAIA), resembles actively induced CIA, with a much more rapid onset (24–48 h), but in acute form (Figure 1). LPS (ligand for toll-like receptors, TLR4/TLR2) [41,46] or lipomannan (ligand for TLR2) [47] enhances the incidence and severity of the antibody initiated disease by decreasing the threshold for arthritis induction. Disease susceptibility is independent of MHC alleles [27,42] and severe combined immunodeficient (SCID) mice developed arthritis [48], as well as T or B cell deficient mice [49], but the T and B cell double deficient mice had less severe arthritis [49], suggesting a regulatory role for these cells at the effector level [50,51,52]. CAIA is an acute arthritis that is triggered by antibody binding and neutrophils/macrophages, but bypassing the adaptive immune responses.
For CAIA induction, IL-1β, TNF-α and MIP-1α are required, but not IL-6 [48]. IL-4 [53,54] and IL-10 [55] promoted the disease. Several complement components and their receptors [28,56,57,58,59,60,61] are involved. The complement factor 5 (C5) break down product, C5a is the most potent anaphylatoxin and a powerful chemo-attractant for neutrophils and monocytes, with the ability to promote margination, extravasation, and activation of these cells [62]. C5a levels are markedly elevated in the synovial fluids of patients with RA [63], and a selective C5a receptor antagonist is inhibitory to immune complex–induced inflammation [64]. Hence, C5a plays a crucial role in antibody mediated arthritis [65] and a recombinant vaccine, which induced C5a-specific neutralizing antibodies attenuated CAIA development [66]. Similarly, a fusion protein containing synovial-homing peptide and anti-C5 neutralizing antibody, which specifically targeted inflamed joints attenuated antibody initiated arthritis [67]. Presumably, inflammatory cell recruitment to the joint by C5a or by other complement-induced chemotactic factors are required for the disease initiation.
Interestingly, C5a binding to C5aR induces the expression of activating FcγRIII, while down modulating inhibitory FcγRII on macrophages, which demonstrates how these two key components of acute inflammation can interact with each other in vivo [68]. Mice lacking the common γ-chain of FcRs are highly resistant [45,69] to CAIA, but are only partially resistant in FcγRIII deficient mice [69]. The absence of FcγRII in DBA/1 mice exacerbates the disease [45], but not so in the BALB/c background [69]. More rapid and severe arthritis was observed with an injection of single anti-CII antibody in FcγIIa transgenic mice [70]. Recent observations also highlight the difference in effector functions of IgG Fc engaged to the complement components and FcγRs [71]. There are several factors that could influence the relative contributions of complement versus FcR dependent inflammatory pathways to the immune complex-triggered inflammatory responses. These include antibody isotype, titer as well as the site of immune complex deposition. With respect to Ig isotype, FcR mechanisms could predominate with immune complexes comprised of non-complement-fixing antibodies or after deposition in sites with abundant resident FcR-bearing inflammatory cells. Conversely, complement-driven inflammation may dominate when immune complexes containing Ig-constant regions poorly bound by FcR or when leukocytes must be attracted to an inflammatory site. In addition, antibody titer may influence the humoral pathways of inflammation [72] and subsequent antibody synthesis by feedback regulation [73]. It has also been shown that C5a can down modulate TLR4 induced immune responses [74], indicating the complexity of interactions occurring during antibody initiated inflammation. In essence, IgG mediated inflammation is mainly dependent on age, sex, FcγRs, complement factors, cytokines (IL-1β, IL-4, IL-10, TNF-α, IFN-β and -γ), chemokines, neutrophils, macrophages, different types of proteases, and other inflammatory mediators, like prostaglandins, leukotrienes, etc. [75,76,77] (Figure 2).
Interestingly, apart from the above described inflammatory phase, antibodies could be pathogenic to the cartilage independent of inflammatory cells and factors [78]. Anti-CII antibodies could be pathogenic to chondrocytes, even in the absence of inflammatory mediators, like involvement in impaired cartilage formation [79], strong inhibition of collagen fibrillogenesis [80], and disorganization of CII fibrils in the extracellular matrix (ECM) with or without increased matrix synthesis [81]. In addition, these pathogenic monoclonal antibodies (mAbs) also induce deleterious effects on cartilage [82,83,84] and inhibit CII self-assembly, which suggests that pathogenic antibodies could possibly interfere with the crucial epitopes at sites essential for the stabilization of the polymeric CII fibrils, leading to disturbances in the integrity of the cartilage matrix. Hence, it is plausible that autoantibodies after binding to the cartilage could initiate unwinding of the triple helical structure of CII, which in turn could lead to proteoglycan depletion [85], allowing more enzymes, inflammatory cells to penetrate into the cartilage architecture to induce further damage. But, direct evidence for these suggested initial pathological events is still not available. Surprisingly, instead of LPS or lipomannan, when mannan from Saccharomyces cerevisiae was used as the secondary stimulus after anti-CII antibodies transfer, chronic arthritis phenotype developed in mice having low levels of reactive oxygen species [86] suggesting that under certain in vivo conditions, antibodies could also contribute to chronic disease manifestations and disease relapses in RA.
3. COMP-Specific Antibodies
Cartilage oligomeric matrix protein (COMP) is a structural cartilage protein synthesized by chondrocytes and composed of 5 identical subunits, with disulfide bonds near the N-terminal, and with a globular domain at the C-terminal end [87,88]. Immunization with COMP leads to induction of arthritis in rats [89] and mice [90]. Polyclonal antibodies binding to COMP upon passive transfer induced arthritis, albeit at a lower level of severity [90] as compared to CAIA. Subsequently, mAbs to COMP were generated and shown to induce arthritis in mice [91]. In addition, anti-COMP mAbs enhanced arthritis when co-administered with a sub-arthritogenic dose of CII-specific mAb [91].
4. Anti-GPI Antibodies
The F1 progeny (KBN) of the KRN TCR (recognizing bovine RNase presented by Ak) transgenic mice and the non-obese diabetic (NOD) mice carrying MHC class II allele Aβg7 spontaneously developed severe peripheral arthritis beginning at about three weeks of age [92]. T and B cell autoimmunity to the ubiquitous glycolytic enzyme glucose-6-phosphate isomerase (GPI) is the deriving force in this disease model [93]. The KRN TCR recognizes a peptide derived from GPI (residues 282–294), in the context of Aβg7 [94]. After the initiation, the disease proceeds due to the presence of high levels of anti-GPI antibodies that are present in the KBN serum. Injection of recombinant hGPI [95] or hG6PI (325–339) peptide [96] induced arthritis in mice.
Naïve mice injected with KBN serum [97], affinity-purified anti-GPI antibodies [93], or a combination of two or more anti-GPI mAbs [98] induced arthritis. Purified anti-GPI antibodies transferred into the mice localized specifically to distal joints in the front and rear limbs within minutes of injection, saturated within 20 min and remained localized for at least 24 h [99], and the accumulation of immune complexes seems to be possible due to a lack of decay-accelerating factor (DAF) in this tissue [100] and caused macromolecular vasopermeability localized to joints, thus augmenting its severity [101]. The predominant isotype of the antibodies that are present in the KBN serum is γ1 and severe arthritis is maintained if repeated injections of serum are given [97]. Degranulation of mast cells was apparent within an hour [102] and an influx of neutrophils was prominent within 1–2 days [103]; synovial hyperplasia and mononuclear cell infiltration, with pannus formation and erosions of bone and cartilage, began within a week [97,103].
Similar to CAIA, arthritis caused by KBN serum transfer is MHC independent. Also, T and B cells are not required since arthritis developed in recombination activating gene 1 (RAG1) deficient mice [97] but IL-17-producing T cells can augment this autoantibody-induced arthritis [104]. A single injection of anti-GPI antibody caused prolonged and more severe arthritis in B cell-deficient KBN mice [97]. Mice depleted of neutrophils using anti-Gr-1 antibodies are resistant [103] and neutrophil FcγR, C5aR, and CD11a/LFA-1 are critical components [105]. Interestingly, CpG-oligodeoxynucleotides induced cross talk between CD8α+ dendritic cells and NK cells, which resulted in the suppression of neutrophil recruitment to the joint [106]; mice lacking macrophage-like synoviocytes (op/op) are not susceptible [107]. Similarly, mice that were depleted of macrophages by clodronate liposomes were completely resistant. Reconstituting these mice with macrophages from naive animals reversed this resistance [108]. Intravenous immunoglobulins (IVIG) induced expression of FcγRIIB in macrophages but not in neutrophils protected the mice from the disease [107]. Mice having mutations in the stem cell factor (SCF) receptor, c-kit (W/Wv) or its ligand, SCF (Sl/Sld), leading to mast cells deficiency, are resistant, and susceptibility can be restored by reconstitution with mast cell precursors [102,109]. Subsequently, it was shown that mast cells contribute to the antibody initiated arthritis via IL-1 [110]. TNF-α and IL-1R, but not IL-6 deficient mice were resistant to disease induction [111,112], but TNFR1 and TNFR2 deficient mice were susceptible [113]. IL-4 is dispensable [114] and a genetic polymorphism in IL-1β gene was shown to be of importance [115]. Gene-disrupted or congenic mice were used to delineate the roles of complement components: factor B, C3, C5 and C5aR are essential, but not C1q, C4, MBL-1, C6, CR1, 2, and 3 [113]. Thus, it has been concluded that activation through the alternative pathway leading to the generation of C5a is important in the serum transfer arthritis. Mice lacking the common γ-chain of FcRs are more resistant than those lacking only FcγRIII [113]. But, different results were obtained with FcγRII deficient mice, either they had no effect [113], or they had an earlier onset and greater severity of disease [109]. The neonatal MHC-like FcR (FcRn), associated with the half-life of transferred antibodies, is required [116]. NKT cells promoted this antibody-mediated inflammation [117]. Interestingly, IVIG treatment or ant-murine albumin antibodies protected mice against KBN serum induced arthritis [118], suggesting the importance of antibody-FcR interactions in arthritis pathogenesis.
5. Immune-Complex Mediated Arthritis
Immune-complex arthritis (ICA) was elicited in naive mice using a non-self-antigen [119]. Mice injected intravenously with heat-inactivated polyclonal rabbit anti-lysozyme serum, followed by an injection with poly-L-lysine-coupled lysozyme in the joint developed arthritis. Disease featuring a massive influx of neutrophils is evident within a day and wanes over the course of a week. Antigen is deposited on the articular surface, presumably in complex with specific antibody [119]. Local depletion of macrophage-like synoviocytes prevents disease [120]. IL-1 is required for inflammation and cartilage destruction, but TNF-α may be dispensable. In this model, FcγRIII is required for inflammation and cartilage breakdown, but FcγRI seems to be only important in cartilage loss [121], whereas IFN-γ bypasses the dependence on FcγRIII [122]. FcγRII plays a suppressive role, since inflammation and cartilage breakdown are enhanced in FcγRII deficient mice [121]. Chondrocyte death in FcγRI−/− mice was completely abrogated, whereas matrix metalloproteinases (MMPs) mediated cartilage destruction was significantly diminished [121]. Local adenoviral overexpression of IFN-γ in the knee joint prior to the onset of IC-mediated arthritis aggravated severe cartilage destruction. IFN-γ stimulated ICA showed pronounced chondrocyte death that was also completely mediated by FcγRI [123]. Thus, during ICA, synovial macrophages seem to be the dominant factor in the induction of severe cartilage destruction [124].
6. Anti-Citrullinated Peptide/Protein Antibodies
Several citrullinated autoantigens (α-enolase, fibrinogen, filaggrin, vimentin, and CII) are used as targets of ACPAs in the diagnostic assays [125]. Around 70% of RA patients sera contain antibodies binding to cyclic citrullinated peptides (CCP2), and these ACPAs are reported to be associated with more severe arthritis [126]. ACPAs are included as one of the classification criteria for RA by American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) consortium [127]. ACPAs are present in the RA sera decades before the onset of clinical disease [128], possibly suggesting that the triggering for autoimmunity may occur at other locations in the body than the joints [129]. Prior to arthritis onset, epitope spreading [130], avidity maturation [131], and changes towards a pro-inflammatory Fc glycosylation phenotype [132] occurs in the ACPA response.
ACPAs activate osteoclasts [133], leading to bone loss even before the onset of clinical disease [134] and the glycosylation status of IgG determines osteoclast differentiation and bone loss [135]. Thus, autoantibodies could have direct influence on osteoclastogenesis by binding to certain activating FcγRs present on immature osteoclasts leading to enhanced osteoclast generation and bone destruction [136]. Binding of ACPAs to osteoclasts releases IL-8, leading to bone erosion [137] and pain [138], which in turn, could lead to pro-inflammatory processes [139]. Furthermore, ACPAs induce macrophages to secrete TNF-α, mediate activation of complement cascades [140], and FcγRIIa-dependent activation of platelets [141]. ACPAs are also shown to be pathogenic in experimental arthritis [142,143]. Hence, it is plausible that ACPAs may play a crucial part in RA pathogenesis [144].
7. Antibody Induced Pain
Autoantibodies binding to target tissues can induce pain through Fc, Fab-dependent mechanisms [145] possibly via inflammatory mediators like high mobility group box-1 protein (HMGB1) [146] or chemokines released from osteoclasts [138]. Arthralgia in RA patients’ may precede joint inflammation, may not correlate with the degree of inflammation, and may persist even after successful treatment of inflammation. In this context, KBN serum transfer induced persistent pain and TNF-α/prostaglandin inhibitors attenuated the allodynia induced during inflammation [147]. Experiments with CII-specific pathogenic IgG antibodies demonstrated time-dependent prostaglandin and spinal glial contribution to antibody-induced pain [148]. Spinal HMGB1 also contributes to nociceptive signal transmission via the activation of TLR4 in antibody induced inflammation [146].
8. Protective Autoantibodies
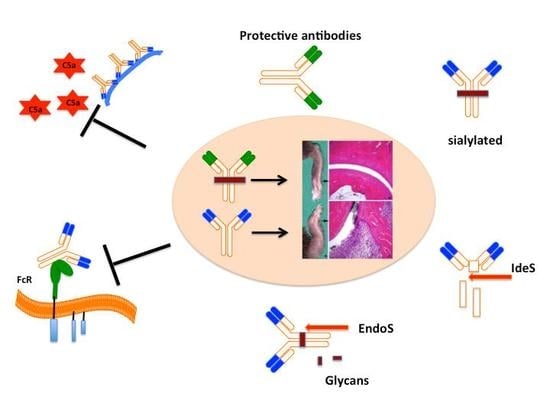
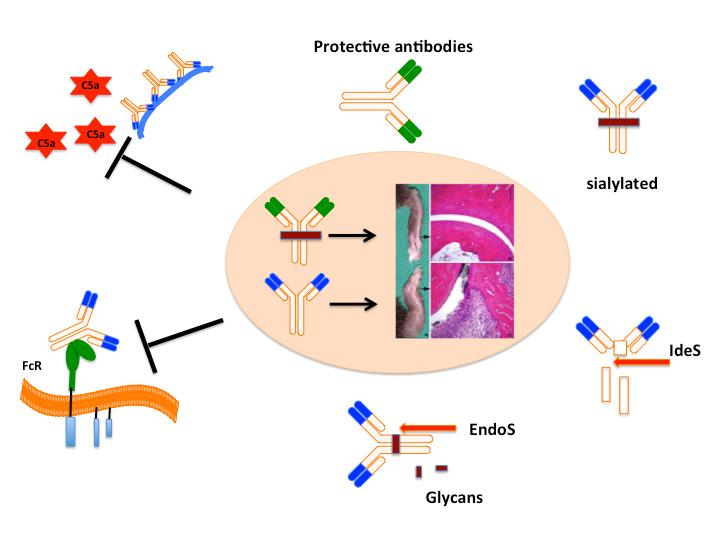
Interestingly, not all the antibodies are pathogenic in nature. Some of them could be protective, which suggests the possible regulation at the effector level of arthritis. One of the anti-CII antibodies, named CIIF4 binding to the CII epitope, F4 (ERGLKGHRGFT, amino acids Gly926-Phe936) has a protective role against arthritis, when given in combination with arthritogenic antibodies [85,149]. Cartilage explant studies showed that CIIF4 penetrated the extracellular matrix during culture, remained bound to the tissue [82], induced negligible loss of proteoglycan, minimal chemical changes in the composition of the matrix [85], and allowed matrix regeneration, which required viable chondrocytes [150]. Similarly, one of the ACPA mAbs binding to citrullinated fibrinogen [132,151] was found to be protective [152]. However, the mechanisms (for example, steric hindrance for pathogenic antibody binding to the cartilage, blocking of MMP cleavage sites and/or having protective IgG N-glycome profile) of antibody protection are still not clear.
9. Targeting IgG to Treat Antibody Dependent Pathologies
At the effector level of arthritis, apart from targeting effector molecules, like C5 [67,153] and its break down product C5a [65,154,155], receptors (FcRs [156], TLRs [157]), transcription factors [158,159]), and cytokines, using different strategies and drugs [160,161,162], methods for direct targeting of pathogenic IgG antibodies could be attractive and optimal for therapeutic applications.
IgG molecules at Asn-297 of the CH2 domain of IgG Fc part are glycosylated with variable galactosylation and limited sialylation [163]. Changes in N-glycome alter Fc conformation with direct effects on IgG effector functions [164,165] and have important immunoregulatory functions [166]. For example, increasing afucosylated glycoforms by glyco-engineering have significantly increased the cell mediated cytotoxicity of the target bound anti-CD20 antibody [167]. It is clear that sialylation of the of the Fc fragment confers anti-inflammatory properties [168,169]. Anti-inflammatory property of intravenous IgGs (IVIGs) is mainly attributed to sialylated glycans present in the Fc part of IgG [169,170]. Abrogation of the arthritis activity of KBN sera was observed when sialic acids attached to the penultimate galactose of IgG Fc by α2,6 linkages were cleaved using sialidase or after administration of sialic acid enriched Fc fragments [171]. Sialylated Fcs bind to a specific C-type lectin receptors, SIGN-R1 expressed on macrophages [172], leading to the up-regulation of the inhibitory FcγRIIb on inflammatory cells and inhibition of autoantibody initiated inflammation [173,174] via production of IL-33 and, IL-4 [175] acting on IL-4α [176]. Interestingly, sialylation of anti-CII antibodies and ACPAs attenuates arthritogenic activity and leads to suppression of CIA [177]. Recently, several methods have been developed to modulate the glycan pattern of an antibody for therapeutic benefits (for recent review, see [178]).
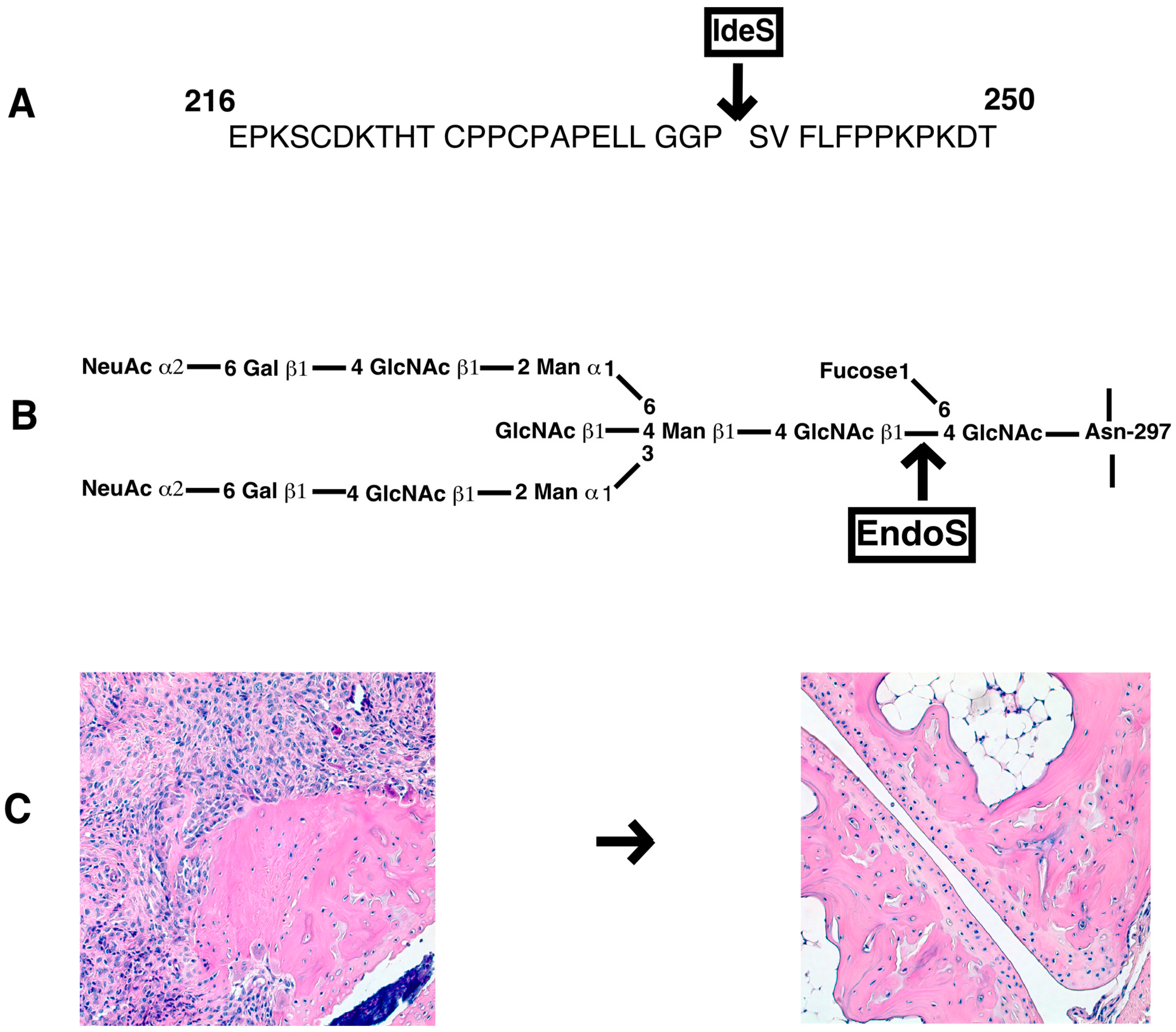
Bacterial enzymes to specifically cleave IgG at the hinge region or remove specific carbohydrate moieties linked to the N-glycans of the Fc core polysaccharides could also be used for inhibition of antibody induced inflammation (Figure 3). Endo-β-N-acetylglucosaminidase (EndoS) is a member of the GlcNAc polymer hydrolyzing glycosyl hydrolases of family 18-glycosyl hydrolase secreted by group A β-hemolytic Streptococcus pyogenes. It exclusively hydrolyses the β-1,4-di-N-acetylchitobiose core of the N-linked complex type glycan on Asn-297 of the γ-chains of IgG [179]. EndoS treatment of antibodies did not affect binding of IgG to CII and complement activation, but reduced binding to FcγRs and formation of stable immune complexes [180]. EndoS treatment of KBN serum decreased inflammation induced by anti-GPI antibodies [181]. Similarly, pathogenic potential of IgG molecules were attenuated in other inflammatory conditions as well [182]. EndoS is extremely potent in disrupting larger immune complex lattice formation on the cartilage surface possibly through the destabilization of Fc-Fc interactions [183]. Treatment of mice with EndoS has suppressed many antibody mediated experimental autoimmune diseases (thrombocytopenic purpura [184], arthritis [181], glomerulonephritis [185,186], encephalomyelitis [187], hemolytic anemia [188], and epidermolysis bullosa acquisita [189]). Recent studies also showed that treatment with EndoS reduced Fc/FcγR interactions through Fc deglycosylation, which led to reduction in immune complex-mediated neutrophil activation [190].
Another enzyme secreted by S. pyogenes is the IgG-degrading enzyme (IdeS), a cysteine endopeptidase, which cleaves the heavy chains of IgG with a unique specificity [191]. By removing the Fc part from the antigen recognizing Fab, immune responses such as complement activation and Fc dependent effector mechanisms are eliminated. IdeS completely blocked antibody-induced arthritis, reduced CIA disease severity, and inhibited antibody initiated arthritis relapses [192]. Similarly, IdeS is effective in ameliorating other IgG dependent pathologies [182]. Recently, IdeS was shown to reduce/eliminate donor specific antibodies and permitted HLA-incompatible transplantation in patients [193]. Interestingly, IdeS can also cleave IgG type B cell receptors, leading to abolished receptor mediated signal transduction and memory B cell activation, temporarily [194].
Thus, glyco-engineering of IgG molecules [195], use of bacterial enzymes to specifically cleave IgG or remove certain carbohydrate moieties [78,182], or blocking the downstream effector pathways [65] to ameliorate IgG dependent pathologies offer new avenues for novel drug development. It is of interest to note that several modifications have been reported that could modulate the therapeutic capability of IgG antibodies [196] and designing of antibodies for improving their therapeutic potency has been reviewed recently [197].
10. Conclusions
At the IgG mediated effector level of arthritis, different pathways of complement activation, FcγR engagement, activation of residential, and infiltrated immune cells in the synovia, various cytokine and chemokine secretion are essential for the development of clinical disease. Requirement for the APC derived cytokines, TNF-α and IL-1β for arthritis induction and perpetuation is obvious. Whereas, T cell secreted cytokines could be detrimental or protective to the joints, depending on the phase of the clinical disease and in situ conditions. Effector cells of the innate immune system (neutrophils, macrophages, and mast cells) drawn to the inflammatory foci by different chemokines and chemo-attractants are actively engaged to induce inflammation, inflict damage to the cartilage, and perpetuate the ongoing immune responses by secreting cytokines and proteases. Once the stimuli (pathogenic IgG molecules) are eliminated, the inflammation subsides. However, if epitope spreading and release of unexposed antigens or antigenic modifications in the presence of strong immune stimuli (for example, mannan) are continuing within the joint, it could drive the acute disease into chronic inflammation under certain conditions with a complete disruption of joint architecture. Hence, it would be valuable to dissect the fine specificity of the molecules taking part in the pathogenesis, as well as understanding both the upstream and downstream molecular events that are involved in the antibody mediated disease process for effective development of therapeutic strategies. With the recent advances in our knowledge and techniques in various scientific disciplines, the possibility of developing such novel therapies for RA is all the more promising.
Acknowledgments
The author acknowledges the project grant from Southern Medical University, Guangzhou, China (No. C1034211).
Conflicts of Interest
The author declares no conflict of interest.
References
- Orr, C.; Vieira-Sousa, E.; Boyle, D.L.; Buch, M.H.; Buckley, C.D.; Cañete, J.D.; Catrina, A.I.; Choy, E.H.S.; Emery, P.; Fearon, U.; et al. Synovial tissue research: A state-of-the-art review. Nat. Rev. Rheumatol. 2017, 13, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.-S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Buckley, C.D.; Isaacs, J.D. Cytokines in rheumatoid arthritis—Shaping the immunological landscape. Nat. Rev. Rheumatol. 2016, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Chan, A.C. B cell immunobiology in disease: Evolving concepts from the clinic. Annu. Rev. Immunol. 2006, 24, 467–496. [Google Scholar] [CrossRef] [PubMed]
- Steiner, G.; Smolen, J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res. 2002, 4 (Suppl. 2), S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yang, F.; Huang, C.; Huang, J.; Wang, Q.; Chen, Y.; Du, Y.; Zhao, L.; Gao, M.; Wang, F. Plasmapheresis therapy in combination with TNF-α inhibitor and DMARDs: A multitarget method for the treatment of rheumatoid arthritis. Mod. Rheumatol. 2017, 27, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.L.; Takase-Minegishi, K.; Ramiro, S.; Chatzidionysiou, K.; Smolen, J.S.; van der Heijde, D.; Bijlsma, J.W.; Burmester, G.R.; Dougados, M.; Scholte-Voshaar, M.; et al. Efficacy of biological disease-modifying antirheumatic drugs: A systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1113–1136. [Google Scholar] [CrossRef] [PubMed]
- Holmdahl, R.; Andersson, M.; Goldschmidt, T.J.; Gustafsson, K.; Jansson, L.; Mo, J.A. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol. Rev. 1990, 118, 193–232. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Jirholt, J.; Holmdahl, R.; Jansson, L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin. Exp. Immunol. 1998, 111, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A.; Johansson, A.; Vestberg, M.; Holmdahl, R. Collagen-induced arthritis development requires alpha beta T cells but not gamma delta T cells: Studies with T cell-deficient (TCR mutant) mice. Int. Immunol. 1999, 11, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Wooley, P.H.; Luthra, H.S.; Griffiths, M.M.; Stuart, J.M.; Huse, A.; David, C.S. Type II collagen-induced arthritis in mice. IV. Variations in immunogenetic regulation provide evidence for multiple arthritogenic epitopes on the collagen molecule. J. Immunol. 1985, 135, 2443–2451. [Google Scholar] [PubMed]
- Nabozny, G.H.; Baisch, J.M.; Cheng, S.; Cosgrove, D.; Griffiths, M.M.; Luthra, H.S.; David, C.S. HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: A novel model for human polyarthritis. J. Exp. Med. 1996, 183, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rosloniec, E.F.; Brand, D.D.; Myers, L.K.; Whittington, K.B.; Gumanovskaya, M.; Zaller, D.M.; Woods, A.; Altmann, D.M.; Stuart, J.M.; Kang, A.H. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J. Exp. Med. 1997, 185, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.C.; Hansen, B.E.; Jacobsen, H.; Madsen, L.S.; Andersen, C.B.; Engberg, J.; Rothbard, J.B.; McDevitt, G.S.; Malmström, V.; Holmdahl, R.; et al. Definition of MHC and T cell receptor contacts in the HLA-DR4restricted immunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7574–7579. [Google Scholar] [CrossRef] [PubMed]
- Michaëlsson, E.; Andersson, M.; Engström, A.; Holmdahl, R. Identification of an immunodominant type-II collagen peptide recognized by T cells in H-2q mice: Self tolerance at the level of determinant selection. Eur. J. Immunol. 1992, 22, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Rosloniec, E.F.; Whittington, K.B.; Brand, D.D.; Myers, L.K.; Stuart, J.M. Identification of MHC class II and TCR binding residues in the type II collagen immunodominant determinant mediating collagen-induced arthritis. Cell. Immunol. 1996, 172, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lambert, L.E.; Berling, J.S. Structural requirements for recognition of a type II collagen peptide by murine T cell hybridomas. Cell. Immunol. 1994, 153, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Michaëlsson, E.; Broddefalk, J.; Engström, A.; Kihlberg, J.; Holmdahl, R. Antigen processing and presentation of a naturally glycosylated protein elicits major histocompatibility complex class II-restricted, carbohydrate-specific T cells. Eur. J. Immunol. 1996, 26, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Michaëlsson, E.; Malmström, V.; Reis, S.; Engström, A.; Burkhardt, H.; Holmdahl, R. T cell recognition of carbohydrates on type II collagen. J. Exp. Med. 1994, 180, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A.; Bäcklund, J.; Broddefalk, J.; Michaëlsson, E.; Goldschmidt, T.J.; Kihlberg, J.; Holmdahl, R. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur. J. Immunol. 1998, 28, 2580–2590. [Google Scholar] [CrossRef]
- Mo, J.A.; Bona, C.A.; Holmdahl, R. Variable region gene selection of immunoglobulin G-expressing B cells with specificity for a defined epitope on type II collagen. Eur. J. Immunol. 1993, 23, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Khmaladze, I.; Jia, H.; Bajtner, E.; Nandakumar, K.S.; Blom, T.; Mo, J.A.; Holmdahl, R. Pathogenic autoreactive B cells are not negatively selected toward matrix protein collagen II. J. Immunol. 2011, 187, 4451–4458. [Google Scholar] [CrossRef] [PubMed]
- Trentham, D.E.; Townes, A.S.; Kang, A.H. Autoimmunity to type II collagen an experimental model of arthritis. J. Exp. Med. 1977, 146, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Terato, K.; Hasty, K.A.; Cremer, M.A.; Stuart, J.M.; Townes, A.S.; Kang, A.H. Collagen-induced arthritis in mice. Localization of an arthritogenic determinant to a fragment of the type II collagen molecule. J. Exp. Med. 1985, 162, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Dixon, F.J. Serum transfer of collagen-induced arthritis in mice. J. Exp. Med. 1983, 158, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kristan, J.; Hao, L.; Lenkoski, C.S.; Shen, Y.; Matis, L.A. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J. Immunol. 2000, 164, 4340–4347. [Google Scholar] [CrossRef] [PubMed]
- Bajtner, E.; Nandakumar, K.S.; Engström, A.; Holmdahl, R. Chronic development of collagen-induced arthritis is associated with arthritogenic antibodies against specific epitopes on type II collagen. Arthritis Res. Ther. 2005, 7, R1148–R1157. [Google Scholar] [CrossRef] [PubMed]
- Lindh, I.; Snir, O.; Lönnblom, E.; Uysal, H.; Andersson, I.; Nandakumar, K.S.; Vierboom, M.; ’t Hart, B.; Malmström, V.; Holmdahl, R. Type II collagen antibody response is enriched in the synovial fluid of rheumatoid joints and directed to the same major epitopes as in collagen induced arthritis in primates and mice. Arthritis Res. Ther. 2014, 16, R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plows, D.; Kontogeorgos, G.; Kollias, G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J. Immunol. 1999, 162, 1018–1023. [Google Scholar] [PubMed]
- Mo, J.A.; Holmdahl, R. The B cell response to autologous type II collagen: Biased V gene repertoire with V gene sharing and epitope shift. J. Immunol. 1996, 157, 2440–2448. [Google Scholar] [PubMed]
- Nandakumar, K.S.; Lindqvist, A.-K.B.; Holmdahl, R. A dominant suppressive MHC class II haplotype interacting with autosomal genes controls autoantibody production and chronicity of arthritis. Ann. Rheum. Dis. 2011, 70, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Förster, M.; Raposo, B.; Ekman, D.; Klaczkowska, D.; Popovic, M.; Nandakumar, K.S.; Lindvall, T.; Hultqvist, M.; Teneva, I.; Johannesson, M.; et al. Genetic control of antibody production during collagen-induced arthritis development in heterogeneous stock mice. Arthritis Rheum. 2012, 64, 3594–3603. [Google Scholar] [CrossRef] [PubMed]
- Raposo, B.; Dobritzsch, D.; Ge, C.; Ekman, D.; Xu, B.; Lindh, I.; Förster, M.; Uysal, H.; Nandakumar, K.S.; Schneider, G.; et al. Epitope-specific antibody response is controlled by immunoglobulin V(H) polymorphisms. J. Exp. Med. 2014, 211, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Holmdahl, R.; Jansson, L.; Larsson, A.; Jonsson, R. Arthritis in DBA/1 mice induced with passively transferred type II collagen immune serum. Immunohistopathology and serum levels of anti-type II collagen auto-antibodies. Scand. J. Immunol. 1990, 31, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Wooley, P.H.; Luthra, H.S.; Singh, S.K.; Huse, A.R.; Stuart, J.M.; David, C.S. Passive transfer of arthritis to mice by injection of human anti-type II collagen antibody. Mayo Clin. Proc. 1984, 59, 737–743. [Google Scholar] [CrossRef]
- Petkova, S.B.; Konstantinov, K.N.; Sproule, T.J.; Lyons, B.L.; Awwami, M.A.; Roopenian, D.C. Human antibodies induce arthritis in mice deficient in the low-affinity inhibitory IgG receptor Fc gamma RIIB. J. Exp. Med. 2006, 203, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Terato, K.; Hasty, K.A.; Reife, R.A.; Cremer, M.A.; Kang, A.H.; Stuart, J.M. Induction of arthritis with monoclonal antibodies to collagen. J. Immunol. 1992, 148, 2103–2108. [Google Scholar] [PubMed]
- Terato, K.; Harper, D.S.; Griffiths, M.M.; Hasty, D.L.; Ye, X.J.; Cremer, M.A.; Seyer, J.M. Collagen-induced arthritis in mice: Synergistic effect of E. coli lipopolysaccharide bypasses epitope specificity in the induction of arthritis with monoclonal antibodies to type II collagen. Autoimmunity 1995, 22, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Svensson, L.; Holmdahl, R. Collagen type II-specific monoclonal antibody-induced arthritis in mice: Description of the disease and the influence of age, sex, and genes. Am. J. Pathol. 2003, 163, 1827–1837. [Google Scholar] [CrossRef]
- Nandakumar, K.S.; Holmdahl, R. Efficient promotion of collagen antibody induced arthritis (CAIA) using four monoclonal antibodies specific for the major epitopes recognized in both collagen induced arthritis and rheumatoid arthritis. J. Immunol. Methods 2005, 304, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Hutamekalin, P.; Saito, T.; Yamaki, K.; Mizutani, N.; Brand, D.D.; Waritani, T.; Terato, K.; Yoshino, S. Collagen antibody-induced arthritis in mice: Development of a new arthritogenic 5-clone cocktail of monoclonal anti-type II collagen antibodies. J. Immunol. Methods 2009, 343, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Andrén, M.; Martinsson, P.; Bajtner, E.; Hellström, S.; Holmdahl, R.; Kleinau, S. Induction of arthritis by single monoclonal IgG anti-collagen type II antibodies and enhancement of arthritis in mice lacking inhibitory Fcgamma RIIB. Eur. J. Immunol. 2003, 33, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-K.; Kang, S.-M.; Paik, D.-J.; Kim, J.M.; Youn, J. Essential roles of Toll-like receptor-4 signaling in arthritis induced by type II collagen antibody and LPS. Int. Immunol. 2005, 17, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Kelkka, T.; Hultqvist, M.; Nandakumar, K.S.; Holmdahl, R. Enhancement of antibody-induced arthritis via Toll-like receptor 2 stimulation is regulated by granulocyte reactive oxygen species. Am. J. Pathol. 2012, 181, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kagari, T.; Doi, H.; Shimozato, T. The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J. Immunol. 2002, 169, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Bäcklund, J.; Vestberg, M.; Holmdahl, R. Collagen type II (CII)-specific antibodies induce arthritis in the absence of T or B cells but the arthritis progression is enhanced by CII-reactive T cells. Arthritis Res. Ther. 2004, 6, R544–R550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fathman, J.W.; Lugo-Villarino, G.; Scimone, L.; von Andrian, U.; Dorfman, D.M.; Glimcher, L.H. Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J. Clin. Investig. 2006, 116, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, M.; Nakano, N.; Yonekawa, T.; Shan, L.; Kaise, T.; Kobayashi, T.; Yamashita, K.; Kikkawa, H.; Kinoshita, M. T cells are involved in the development of arthritis induced by anti-type II collagen antibody. Int. Immunopharmacol. 2007, 7, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Oh, Y.; Park, O.; Foss, C.A.; Lim, S.M.; Jo, D.-G.; Na, D.H.; Pomper, M.G.; Lee, K.C.; Lee, S. PEGylated TRAIL ameliorates experimental inflammatory arthritis by regulation of Th17 cells and regulatory T cells. J. Control. Release 2017, 267, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Nandakumar, K.S.; Johansson, A.; Jansson, L.; Holmdahl, R. IL-4-deficient mice develop less acute but more chronic relapsing collagen-induced arthritis. Eur. J. Immunol. 2002, 32, 2944–2953. [Google Scholar] [CrossRef]
- Nandakumar, K.S.; Holmdahl, R. Arthritis induced with cartilage-specific antibodiesis IL-4-dependent. Eur. J. Immunol. 2006, 36, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Hansson, A.S.; Nandakumar, K.S.; Bäcklund, J.; Holmdahl, R. IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J. Immunol. 2001, 167, 3505–3512. [Google Scholar] [CrossRef] [PubMed]
- Hietala, M.A.; Nandakumar, K.S.; Person, L.; Fahlen, S.; Holmdahl, R.; Pekna, M. Complement activation by both classical and alternative pathways is critical for the effector phase of arthritis. Mol. Immunol. 2003, 40, 190. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Thurman, J.M.; Kraus, D.; Wood, A.; Carroll, M.C.; Arend, W.P.; Holers, V.M. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J. Immunol. 2006, 177, 1904–1912. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Levitt, B.; Glogowska, M.J.; Thurman, J.M.; Takahashi, K.; Stahl, G.L.; Tomlinson, S.; Arend, W.P.; Holers, V.M. Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice. J. Immunol. 2009, 183, 5928–5937. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Hyatt, S.; Antonioli, A.H.; White, J.T.; Glogowska, M.; Takahashi, K.; Merkel, T.J.; Stahl, G.L.; Mueller-Ortiz, S.; Wetsel, R.; et al. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J. Immunol. 2012, 188, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Mehta, G.; Ferreira, V.P.; Cortes, C.; Pickering, M.C.; Pangburn, M.K.; Arend, W.P.; Holers, V.M. Essential role of surface-bound complement factor H in controlling immune complex-induced arthritis. J. Immunol. 2013, 190, 3560–3569. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Acharya, S.; Scheinman, R.I.; Mehta, G.; Takahashi, M.; Endo, Y.; Zhou, W.; Farrar, C.A.; Sacks, S.H.; Fujita, T.; et al. Deconstructing the Lectin Pathway in the Pathogenesis of Experimental Inflammatory Arthritis: Essential Role of the Lectin Ficolin B and Mannose-Binding Protein-Associated Serine Protease 2. J. Immunol. 2017, 199, 1835–1845. [Google Scholar] [CrossRef] [PubMed]
- Gerard, C.; Gerard, N.P. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 1994, 12, 775–808. [Google Scholar] [CrossRef] [PubMed]
- Jose, P.J.; Moss, I.K.; Maini, R.N.; Williams, T.J. Measurement of the chemotactic complement fragment C5a in rheumatoid synovial fluids by radioimmunoassay: Role of C5a in the acute inflammatory phase. Ann. Rheum. Dis. 1990, 49, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Heller, T.; Hennecke, M.; Baumann, U.; Gessner, J.E.; zu Vilsendorf, A.M.; Baensch, M.; Boulay, F.; Kola, A.; Klos, A.; Bautsch, W.; et al. Selection of a C5a receptor antagonist from phage libraries attenuating the inflammatory response in immune complex disease and ischemia/reperfusion injury. J. Immunol. 1999, 163, 985–994. [Google Scholar] [PubMed]
- Woodruff, T.M.; Nandakumar, K.S.; Tedesco, F. Inhibiting the C5-C5a receptor axis. Mol. Immunol. 2011, 48, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Jansson, Å.; Xu, B.; Rydell, N.; Blom, A.M. A Recombinant Vaccine Effectively Induces C5a-Specific Neutralizing Antibodies and Prevents Arthritis. PLoS ONE 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Macor, P.; Durigutto, P.; De Maso, L.; Garrovo, C.; Biffi, S.; Cortini, A.; Fischetti, F.; Sblattero, D.; Pitzalis, C.; Marzari, R.; et al. Treatment of experimental arthritis by targeting synovial endothelium with a neutralizing recombinant antibody to C5. Arthritis Rheum. 2012, 64, 2559–2567. [Google Scholar] [CrossRef] [PubMed]
- Shushakova, N.; Skokowa, J.; Schulman, J.; Baumann, U.; Zwirner, J.; Schmidt, R.E.; Gessner, J.E. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J. Clin. Investig. 2002, 110, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Kagari, T.; Tanaka, D.; Doi, H.; Shimozato, T. Essential role of Fc gamma receptors in anti-type II collagen antibody-induced arthritis. J. Immunol. 2003, 170, 4318–4324. [Google Scholar] [CrossRef] [PubMed]
- Tan Sardjono, C.; Mottram, P.L.; van de Velde, N.C.; Powell, M.S.; Power, D.; Slocombe, R.F.; Wicks, I.P.; Campbell, I.K.; McKenzie, S.E.; Brooks, M.; et al. Development of spontaneous multisystem autoimmune disease and hypersensitivity to antibody-induced inflammation in Fcgamma receptor IIa-transgenic mice. Arthritis Rheum. 2005, 52, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Romain, G.; Yan, W.; Watanabe, M.; Charab, W.; Todorova, B.; Lee, J.; Triplett, K.; Donkor, M.; Lungu, O.I.; et al. IgG Fc domains that bind C1q but not effector Fcγ receptors delineate the importance of complement-mediated effector functions. Nat. Immunol. 2017, 18, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Quigg, R.J.; Lim, A.; Haas, M.; Alexander, J.J.; He, C.; Carroll, M.C. Immune complex glomerulonephritis in C4- and C3-deficient mice. Kidney Int. 1998, 53, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hjelm, F.; Carlsson, F.; Getahun, A.; Heyman, B. Antibody-mediated regulation of the immune response. Scand. J. Immunol. 2006, 64, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hawlisch, H.; Belkaid, Y.; Baelder, R.; Hildeman, D.; Gerard, C.; Köhl, J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity 2005, 22, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Holmdahl, R. Antibody-induced arthritis: Disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res. Ther. 2006, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Nandakumar, K.S.; Holmdahl, R. The role of collagen antibodies in mediating arthritis. Mod. Rheumatol. 2008, 18, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S. Pathogenic antibody recognition of cartilage. Cell Tissue Res. 2010, 339, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Holmdahl, R. Therapeutic cleavage of IgG: New avenues for treating inflammation. Trends Immunol. 2008, 29, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Amirahmadi, S.F.; Pho, M.H.; Gray, R.E.; Crombie, D.E.; Whittingham, S.F.; Zuasti, B.B.; van Damme, M.-P.; Rowley, M.J. An arthritogenic monoclonal antibody to type II collagen, CII-C1, impairs cartilage formation by cultured chondrocytes. Immunol. Cell Biol. 2004, 82, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.E.; Seng, N.; Mackay, I.R.; Rowley, M.J. Measurement of antibodies to collagen II by inhibition of collagen fibril formation in vitro. J. Immunol. Methods 2004, 285, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Amirahmadi, S.F.; Whittingham, S.; Crombie, D.E.; Nandakumar, K.S.; Holmdahl, R.; Mackay, I.R.; van Damme, M.-P.; Rowley, M.J. Arthritogenic anti-type II collagen antibodies are pathogenic for cartilage-derived chondrocytes independent of inflammatory cells. Arthritis Rheum. 2005, 52, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Crombie, D.E.; Turer, M.; Zuasti, B.B.; Wood, B.; McNaughton, D.; Nandakumar, K.S.; Holmdahl, R.; van Damme, M.-P.; Rowley, M.J. Destructive effects of murine arthritogenic antibodies to type II collagen on cartilage explants in vitro. Arthritis Res. Ther. 2005, 7, R927–R937. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.M.; Nandakumar, K.S.; Holmdahl, R.; Tobin, M.J.; McNaughton, D.; Rowley, M.J. Chemical changes demonstrated in cartilage by synchrotron infrared microspectroscopy in an antibody-induced murine model of rheumatoid arthritis. J. Biomed. Opt. 2011, 16, 066004. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.M.; Whittingham, S.; McNaughton, D.; Nandakumar, K.S.; Holmdahl, R.; Rowley, M.J. Type II collagen–specific antibodies induce cartilage damage in mice independent of inflammation. Arthritis Rheum. 2013, 65, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Bajtner, E.; Hill, L.; Böhm, B.; Rowley, M.J.; Burkhardt, H.; Holmdahl, R. Arthritogenic antibodies specific for a major type II collagen triple-helical epitope bind and destabilize cartilage independent of inflammation. Arthritis Rheum. 2008, 58, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Hagert, C.; Sareila, O.; Kelkka, T.; Nandakumar, K.S.; Collin, M.; Xu, B.; Guerard, S.; Bäcklund, J.; Jalkanen, S.; Holmdahl, R. Chronic active arthritis driven by macrophages without involvement of T cells. Arthritis Rheum. 2002, 4 (Suppl. 3), S197–S211. [Google Scholar]
- Mörgelin, M.; Heinegård, D.; Engel, J.; Paulsson, M. Electron microscopy of native cartilage oligomeric matrix protein purified from the Swarm rat chondrosarcoma reveals a five-armed structure. J. Biol. Chem. 1992, 267, 6137–6141. [Google Scholar] [PubMed]
- Oldberg, A.; Antonsson, P.; Lindblom, K.; Heinegård, D. COMP (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J. Biol. Chem. 1992, 267, 22346–22350. [Google Scholar] [PubMed]
- Carlsén, S.; Hansson, A.S.; Olsson, H.; Heinegård, D.; Holmdahl, R. Cartilage oligomeric matrix protein (COMP)-induced arthritis in rats. Clin. Exp. Immunol. 1998, 114, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, S.; Nandakumar, K.S.; Bäcklund, J.; Holmberg, J.; Hultqvist, M.; Vestberg, M.; Holmdahl, R. Cartilage oligomeric matrix protein induction of chronic arthritis in mice. Arthritis Rheum. 2008, 58, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Nandakumar, K.S.; Pramhed, A.; Aspberg, A.; Mattsson, R.; Holmdahl, R. Cartilage oligomeric matrix protein specific antibodies are pathogenic. Arthritis Res. Ther. 2012, 14, R191. [Google Scholar] [CrossRef] [PubMed]
- Kouskoff, V.; Korganow, A.S.; Duchatelle, V.; Degott, C.; Benoist, C.; Mathis, D. Organ-specific disease provoked by systemic autoimmunity. Cell 1996, 87, 811–822. [Google Scholar] [CrossRef]
- Matsumoto, I.; Staub, A.; Benoist, C.; Mathis, D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 1999, 286, 1732–1735. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Horvath, S.; Matsumoto, I.; Fremont, D.H.; Allen, P.M. Molecular basis for recognition of an arthritic peptide and a foreign epitope on distinct MHC molecules by a single TCR. J. Immunol. 2000, 164, 5788–5796. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Maier, B.; Morawietz, L.; Krenn, V.; Kamradt, T. Immunization with glucose-6-phosphate isomerase induces T cell-dependent peripheral polyarthritis in genetically unaltered mice. J. Immunol. 2004, 172, 4503–4509. [Google Scholar] [CrossRef] [PubMed]
- Iwanami, K.; Matsumoto, I.; Tanaka, Y.; Inoue, A.; Goto, D.; Ito, S.; Tsutsumi, A.; Sumida, T. Arthritogenic T cell epitope in glucose-6-phosphate isomerase-induced arthritis. Arthritis Res. Ther. 2008, 10, R130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korganow, A.S.; Ji, H.; Mangialaio, S.; Duchatelle, V.; Pelanda, R.; Martin, T.; Degott, C.; Kikutani, H.; Rajewsky, K.; Pasquali, J.L.; et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 1999, 10, 451–461. [Google Scholar] [CrossRef]
- Maccioni, M.; Zeder-Lutz, G.; Huang, H.; Ebel, C.; Gerber, P.; Hergueux, J.; Marchal, P.; Duchatelle, V.; Degott, C.; van Regenmortel, M.; et al. Arthritogenic monoclonal antibodies from K/BxN mice. J. Exp. Med. 2002, 195, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Wipke, B.T.; Wang, Z.; Kim, J.; McCarthy, T.J.; Allen, P.M. Dynamic visualization of a joint-specific autoimmune response through positron emission tomography. Nat. Immunol. 2002, 3, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Wipke, B.T.; Wang, Z.; Nagengast, W.; Reichert, D.E.; Allen, P.M. Staging the initiation of autoantibody-induced arthritis: A critical role for immune complexes. J. Immunol. 2004, 172, 7694–7702. [Google Scholar] [CrossRef] [PubMed]
- Binstadt, B.A.; Patel, P.R.; Alencar, H.; Nigrovic, P.A.; Lee, D.M.; Mahmood, U.; Weissleder, R.; Mathis, D.; Benoist, C. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat. Immunol. 2006, 7, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Friend, D.S.; Gurish, M.F.; Benoist, C.; Mathis, D.; Brenner, M.B. Mast cells: A cellular link between autoantibodies and inflammatory arthritis. Science 2002, 297, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Wipke, B.T.; Allen, P.M. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 2001, 167, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Wu, H.-J.; Benoist, C.; Mathis, D. IL-17-producing T cells can augment autoantibody-induced arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 21789–21794. [Google Scholar] [CrossRef] [PubMed]
- Monach, P.A.; Nigrovic, P.A.; Chen, M.; Hock, H.; Lee, D.M.; Benoist, C.; Mathis, D. Neutrophils in a mouse model of autoantibody-mediated arthritis: Critical producers of Fc receptor gamma, the receptor for C5a, and lymphocyte function-associated antigen 1. Arthritis Rheum. 2010, 62, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Sawaya, H.; Binstadt, B.; Brickelmaier, M.; Blasius, A.; Gorelik, L.; Mahmood, U.; Weissleder, R.; Carulli, J.; Benoist, C.; et al. Inflammatory arthritis can be reined in by CpG-induced DC-NK cell cross talk. J. Exp. Med. 2007, 204, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P.; Samuelsson, A.; Pollard, J.W.; Ravetch, J.V. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity 2003, 18, 573–581. [Google Scholar] [CrossRef]
- Solomon, S.; Rajasekaran, N.; Jeisy-Walder, E.; Snapper, S.B.; Illges, H. A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur. J. Immunol. 2005, 35, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Corr, M.; Crain, B. The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J. Immunol. 2002, 169, 6604–6609. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A.; Binstadt, B.A.; Monach, P.A.; Johnsen, A.; Gurish, M.; Iwakura, Y.; Benoist, C.; Mathis, D.; Lee, D.M. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc. Natl. Acad. Sci. USA 2007, 104, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Pettit, A.; Ohmura, K.; Ortiz-Lopez, A.; Duchatelle, V.; Degott, C.; Gravallese, E.; Mathis, D.; Benoist, C. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J. Exp. Med. 2002, 196, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.-Y.; Crain, B.; Wu, S.R.; Corr, M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J. Exp. Med. 2003, 197, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ohmura, K.; Mahmood, U.; Lee, D.M.; Hofhuis, F.M.A.; Boackle, S.A.; Takahashi, K.; Holers, V.M.; Walport, M.; Gerard, C.; et al. Arthritis critically dependent on innate immune system players. Immunity 2002, 16, 157–168. [Google Scholar] [CrossRef]
- Ohmura, K.; Nguyen, L.T.; Locksley, R.M.; Mathis, D.; Benoist, C. Interleukin-4 can be a key positive regulator of inflammatory arthritis. Arthritis Rheum. 2005, 52, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, K.; Johnsen, A.; Ortiz-Lopez, A.; Desany, P.; Roy, M.; Besse, W.; Rogus, J.; Bogue, M.; Puech, A.; Lathrop, M.; et al. Variation in IL-1beta gene expression is a major determinant of genetic differences in arthritis aggressivity in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 12489–12494. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Petkova, S.; Sproule, T.J.; Shaffer, D.J.; Christianson, G.J.; Roopenian, D. The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J. Clin. Investig. 2004, 113, 1328–1333. [Google Scholar] [PubMed]
- Kim, H.Y.; Kim, H.J.; Min, H.S.; Kim, S.; Park, W.S.; Park, S.H.; Chung, D.H. NKT cells promote antibody-induced joint inflammation by suppressing transforming growth factor beta1 production. J. Exp. Med. 2005, 201, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Siragam, V.; Brinc, D.; Crow, A.R.; Song, S.; Freedman, J.; Lazarus, A.H. Can antibodies with specificity for soluble antigens mimic the therapeutic effects of intravenous IgG in the treatment of autoimmune disease? J. Clin. Investig. 2005, 115, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, P.L.; van den Bersselaar, L.A.; van den Hoek, A.E.; van de Loo, A.A.; van den Berg, W.B. Cationic immune complex arthritis in mice—A new model. Synergistic effect of complement and interleukin-1. Am. J. Pathol. 1992, 140, 1451–1461. [Google Scholar] [PubMed]
- Van Lent, P.L.; van den Hoek, A.E.; van den Bersselaar, L.A.; Spanjaards, M.F.; Van Rooijen, N.; Dijkstra, C.D.; Van de Putte, L.B.; van den Berg, W.B. In vivo role of phagocytic synovial lining cells in onset of experimental arthritis. Am. J. Pathol. 1993, 143, 1226–1237. [Google Scholar] [PubMed]
- Nabbe, K.C.A.M.; Blom, A.B.; Holthuysen, A.E.M.; Boross, P.; Roth, J.; Verbeek, S.; van Lent, P.L.E.M.; van den Berg, W.B. Coordinate expression of activating Fc gamma receptors I and III and inhibiting Fc gamma receptor type II in the determination of joint inflammation and cartilage destruction during immune complex-mediated arthritis. Arthritis Rheum. 2003, 48, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Nabbe, K.C.A.M.; Boross, P.; Holthuysen, A.E.M.; Sloëtjes, A.W.; Kolls, J.K.; Verbeek, S.; van Lent, P.L.E.M.; van den Berg, W.B. Joint inflammation and chondrocyte death become independent of Fcgamma receptor type III by local overexpression of interferon-gamma during immune complex-mediated arthritis. Arthritis Rheum. 2005, 52, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Nabbe, K.C.; van Lent, P.L.; Holthuysen, A.E.; Kolls, J.K.; Verbeek, S.; van den Berg, W.B. FcgammaRI up-regulation induced by local adenoviral-mediated interferon-gamma production aggravates chondrocyte death during immune complex-mediated arthritis. Am. J. Pathol. 2003, 163, 743–752. [Google Scholar] [CrossRef]
- Van Lent, P.L.; Licht, R.; Dijkman, H.; Holthuysen, A.E.; Berden, J.H.; van den Berg, W.B. Uptake of apoptotic leukocytes by synovial lining macrophages inhibits immune complex-mediated arthritis. J. Leukoc. Biol. 2001, 70, 708–714. [Google Scholar] [PubMed]
- Burska, A.N.; Hunt, L.; Boissinot, M.; Strollo, R.; Ryan, B.J.; Vital, E.; Nissim, A.; Winyard, P.G.; Emery, P.; Ponchel, F. Autoantibodies to posttranslational modifications in rheumatoid arthritis. Mediat. Inflamm. 2014, 2014, 492873. [Google Scholar] [CrossRef] [PubMed]
- Van Gaalen, F.A.; Linn-Rasker, S.P.; van Venrooij, W.J.; de Jong, B.A.; Breedveld, F.C.; Verweij, C.L.; Toes, R.E.M.; Huizinga, T.W.J. Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: A prospective cohort study. Arthritis Rheum. 2004, 50, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Malmström, V.; Catrina, A.I.; Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat. Rev. Immunol. 2017, 17, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, D.; Rantapää-Dahlqvist, S.; Ioan-Facsinay, A.; Onnekink, C.; Schwarte, C.M.; Verpoort, K.N.; Drijfhout, J.W.; Huizinga, T.W.J.; Toes, R.E.M.; Pruijn, G.J.M. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann. Rheum. Dis. 2010, 69, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Suwannalai, P.; van de Stadt, L.A.; Radner, H.; Steiner, G.; El-Gabalawy, H.S.; Zijde, C.M.J.-V.D.; van Tol, M.J.; van Schaardenburg, D.; Huizinga, T.W.J.; Toes, R.E.M.; et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, Y.; Ewing, E.; van de Stadt, L.A.; Selman, M.H.J.; Trouw, L.A.; Deelder, A.M.; Huizinga, T.W.J.; Wuhrer, M.; van Schaardenburg, D.; Toes, R.E.M.; et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Kleyer, A.; Finzel, S.; Rech, J.; Manger, B.; Krieter, M.; Faustini, F.; Araujo, E.; Hueber, A.J.; Harre, U.; Engelke, K.; et al. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann. Rheum. Dis. 2014, 73, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Lang, S.C.; Pfeifle, R.; Rombouts, Y.; Frühbeißer, S.; Amara, K.; Bang, H.; Lux, A.; Koeleman, C.A.; Baum, W.; et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat. Commun. 2015, 6, 6651. [Google Scholar] [CrossRef] [PubMed]
- Seeling, M.; Hillenhoff, U.; David, J.P.; Schett, G.; Tuckermann, J.; Lux, A.; Nimmerjahn, F. Inflammatory monocytes and Fcγ receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Haj Hensvold, A.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Bas, D.B.; Fernades-Cerqueira, C.; Krishnamurthy, A.; Nandakumar, K.S.; Rogoz, K.; Kato, J.; Sandor, K.; Su, J.; Jimenez-Andrade, J.M.; et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 2016, 75, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Svensson, C.I.; Malmström, V.; Schett, G.; Klareskog, L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.N.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.J.; Toes, R.E. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009, 60, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Habets, K.L.L.; Trouw, L.A.; Levarht, E.W.N.; Korporaal, S.J.A.; Habets, P.A.M.; de Groot, P.; Huizinga, T.W.J.; Toes, R.E.M. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 209. [Google Scholar] [CrossRef] [PubMed]
- Uysal, H.; Bockermann, R.; Nandakumar, K.S.; Sehnert, B.; Bajtner, E.; Engström, A.; Serre, G.; Burkhardt, H.; Thunnissen, M.M.G.M.; Holmdahl, R. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J. Exp. Med. 2009, 206, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Uysal, H.; Nandakumar, K.S.; Kessel, C.; Haag, S.; Carlsen, S.; Burkhardt, H.; Holmdahl, R. Antibodies to citrullinated proteins: Molecular interactions and arthritogenicity. Immunol. Rev. 2010, 233, 9–33. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Rönnelid, J.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 2008, 26, 651–675. [Google Scholar] [CrossRef] [PubMed]
- Goebel, A. Autoantibody pain. Autoimmun. Rev. 2016, 15, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundbäck, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Christianson, C.A.; Corr, M.; Firestein, G.S.; Mobargha, A.; Yaksh, T.L.; Svensson, C.I. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain 2010, 151, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Bas, D.B.; Su, J.; Sandor, K.; Agalave, N.M.; Lundberg, J.; Codeluppi, S.; Baharpoor, A.; Nandakumar, K.S.; Holmdahl, R.; Svensson, C.I. Collagen antibody-induced arthritis evokes persistent pain with spinal glial involvement and transient prostaglandin dependency. Arthritis Rheum. 2012, 64, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, H.; Koller, T.; Engström, A.; Nandakumar, K.S.; Turnay, J.; Kraetsch, H.G.; Kalden, J.R.; Holmdahl, R. Epitope-specific recognition of type II collagen by rheumatoid arthritis antibodies is shared with recognition by antibodies that are arthritogenic in collagen-induced arthritis in the mouse. Arthritis Rheum. 2002, 46, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.M.; Crombie, D.; McNaughton, D.; Holmdahl, R.; Nandakumar, K.S.; Rowley, M.J. Specific antibody protection of the extracellular cartilage matrix against collagen antibody-induced damage. Arthritis Rheum. 2010, 62, 3374–3384. [Google Scholar] [CrossRef] [PubMed]
- Van de Stadt, L.A.; van Schouwenburg, P.A.; Bryde, S.; Kruithof, S.; van Schaardenburg, D.; Hamann, D.; Wolbink, G.; Rispens, T. Monoclonal anti-citrullinated protein antibodies selected on citrullinated fibrinogen have distinct targets with different cross-reactivity patterns. Rheumatology 2013, 52, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Xu, B.; liang, B.; Nandakumar, K.S.; Tong, D.; Lundqvist, C.; Urbonaviciute, V.; Lönnblom, E.; Ayoglu, B.; Nilsson, P.; et al. Autoantibodies specific for a citrulline side chain protect against arthritis. Ann. Rheum. Dis. 2018. in review. [Google Scholar]
- Durigutto, P.; Macor, P.; Ziller, F.; De Maso, L.; Fischetti, F.; Marzari, R.; Sblattero, D.; Tedesco, F. Prevention of arthritis by locally synthesized recombinant antibody neutralizing complement component C5. PLoS ONE 2013, 8, e58696. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Jansson, A.; Xu, B.; Rydell, N.; Ahooghalandari, P.; Hellman, L.; Blom, A.M.; Holmdahl, R. A recombinant vaccine effectively induces c5a-specific neutralizing antibodies and prevents arthritis. PLoS ONE 2010, 5, e13511. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Scheinman, R.I.; Holers, V.M.; Banda, N.K. A New Approach for the Treatment of Arthritis in Mice with a Novel Conjugate of an Anti-C5aR1 Antibody and C5 Small Interfering RNA. J. Immunol. 2015, 194, 5446–5454. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Xu, Y.; Cen, X.; Nandakumar, K.S.; Liu, S.; Cheng, K. Targeting pattern-recognition receptors to discover new small molecule immune modulators. Eur. J. Med. Chem. 2018, 144, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Kim, D.H.; Nah, S.-S.; Park, M.H.; Lee, H.P.; Han, S.B.; Venkatareddy, U.; Gann, B.; Rodriguez, K.; Burt, S.R.; et al. Novel synthetic (E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) phenol inhibits arthritis by targeting signal transducer and activator of transcription 3. Sci. Rep. 2016, 6, 36852. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Zoheir, K.M.A.; Bakheet, S.A.; Alsaad, A.M.S.; Al-Shabanah, O.A.; Attia, S.M. STA-21, a STAT-3 inhibitor, attenuates the development and progression of inflammation in collagen antibody-induced arthritis. Immunobiology 2017, 222, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Nandakumar, K.S.; Björklund, U.; Holmdahl, R. The novel small molecule drug Rabeximod is effective in reducing disease severity of mouse models of autoimmune disorders. Ann. Rheum. Dis. 2009, 68, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Nandakumar, K.S.; Björklund, U.; Holmdahl, R. Rabeximod reduces arthritis severity in mice by decreasing activation of inflammatory cells. Ann. Rheum. Dis. 2010, 69, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.H.; Lee, W.K.; Lee, Y.S.; Kang, J.S. Assessment of collagen antibody-induced arthritis in BALB/c mice using bioimaging analysis and histopathological examination. Lab. Anim. Res. 2016, 32, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-mediated effector functions: Molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, M.F.; Alter, G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol. 2017, 38, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. The antiinflammatory activity of IgG: The intravenous IgG paradox. J. Exp. Med. 2007, 204, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Schwab, I.; Nimmerjahn, F. Intravenous immunoglobulin therapy: How does IgG modulate the immune system? Nat. Rev. Immunol. 2013, 13, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Nimmerjahn, F.; Ashline, D.J.; Reinhold, V.N.; Paulson, J.C.; Ravetch, J.V. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 2008, 320, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Wermeling, F.; Karlsson, M.C.I.; Ravetch, J.V. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc. Natl. Acad. Sci. USA 2008, 105, 19571–19578. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory actions of intravenous immunoglobulin. Annu. Rev. Immunol. 2008, 26, 513–533. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Ravetch, J.V. A novel role for the IgG Fc glycan: The anti-inflammatory activity of sialylated IgG Fcs. J. Clin. Immunol. 2010, 30 (Suppl. 1), S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Kobayashi, T.; Wermeling, F.; Ravetch, J.V. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature 2011, 475, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Wermeling, F.; Anthony, R.M.; Brombacher, F.; Ravetch, J.V. Acute inflammation primes myeloid effector cells for anti-inflammatory STAT6 signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 13487–13491. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y.; et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat. Commun. 2016, 7, 11205. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; Beck, H.; Rohde, A.; Reusch, D. Therapeutic monoclonal antibody N-glycosylation—Structure, function and therapeutic potential. Biologicals 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Olsén, A. EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J. 2001, 20, 3046–3055. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Collin, M.; Olsén, A.; Nimmerjahn, F.; Blom, A.M.; Ravetch, J.V.; Holmdahl, R. Endoglycosidase treatment abrogates IgG arthritogenicity: Importance of IgG glycosylation in arthritis. Eur. J. Immunol. 2007, 37, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Albert, H.; Collin, M.; Dudziak, D.; Ravetch, J.V.; Nimmerjahn, F. In vivo enzymatic modulation of IgG glycosylation inhibits autoimmune disease in an IgG subclass-dependent manner. Proc. Natl. Acad. Sci. USA 2008, 105, 15005–15009. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Björck, L. Toward Clinical use of the IgG Specific Enzymes IdeS and EndoS against Antibody-Mediated Diseases. Methods Mol. Biol. 2017, 1535, 339–351. [Google Scholar] [PubMed]
- Nandakumar, K.S.; Collin, M.; Happonen, K.E.; Lundstrom, S.L.; Croxford, A.M.; Xu, B.; Zubarev, R.A.; Rowley, M.J.; Blom, A.M.; Kjellman, C.; et al. Streptococcal endo-β-N-acetylglucosaminidase within IgG immune complexes potently suppress inflammation in vivo. Front. Immunol. In review.
- Collin, M.; Shannon, O.; Björck, L. IgG glycan hydrolysis by a bacterial enzyme as a therapy against autoimmune conditions. Proc. Natl. Acad. Sci. USA 2008, 105, 4265–4270. [Google Scholar] [CrossRef] [PubMed]
- Van Timmeren, M.M.; van der Veen, B.S.; Stegeman, C.A.; Petersen, A.H.; Hellmark, T.; Collin, M.; Heeringa, P. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Otten, M.A.; Hellmark, T.; Collin, M.; Björck, L.; Zhao, M.-H.; Daha, M.R.; Segelmark, M. Successful treatment of experimental glomerulonephritis with IdeS and EndoS, IgG-degrading streptococcal enzymes. Nephrol. Dial. Transplant. 2010, 25, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Benkhoucha, M.; Molnarfi, N.; Santiago-Raber, M.-L.; Weber, M.S.; Merkler, D.; Collin, M.; Lalive, P.H. IgG glycan hydrolysis by EndoS inhibits experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2012, 9, 209. [Google Scholar] [CrossRef] [PubMed]
- Allhorn, M.; Briceño, J.G.; Baudino, L.; Lood, C.; Olsson, M.L.; Izui, S.; Collin, M. The IgG-specific endoglycosidase EndoS inhibits both cellular and complement-mediated autoimmune hemolysis. Blood 2010, 115, 5080–5088. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Vafia, K.; Kalies, K.; Groth, S.; Westermann, J.; Zillikens, D.; Ludwig, R.J.; Collin, M.; Schmidt, E. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. J. Autoimmun. 2012, 39, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zheng, J.; Collin, M.; Schmidt, E.; Zillikens, D.; Petersen, F. EndoS reduces the pathogenicity of anti-mCOL7 IgG through reduced binding of immune complexes to neutrophils. PLoS ONE 2014, 9, e85317. [Google Scholar] [CrossRef] [PubMed]
- von Pawel-Rammingen, U.; Johansson, B.P.; Björck, L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J. 2002, 21, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Johansson, B.P.; Björck, L.; Holmdahl, R. Blocking of experimental arthritis by cleavage of IgG antibodies in vivo. Arthritis Rheum. 2007, 56, 3253–3260. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.C.; Lorant, T.; Choi, J.; Kjellman, C.; Winstedt, L.; Bengtsson, M.; Zhang, X.; Eich, T.; Toyoda, M.; Eriksson, B.-M.; et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N. Engl. J. Med. 2017, 377, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Järnum, S.; Bockermann, R.; Runström, A.; Winstedt, L.; Kjellman, C. The Bacterial Enzyme IdeS Cleaves the IgG-Type of B Cell Receptor (BCR), Abolishes BCR-Mediated Cell Signaling, and Inhibits Memory B Cell Activation. J. Immunol. 2015, 195, 5592–5601. [Google Scholar] [CrossRef] [PubMed]
- Pagan, J.D.; Kitaoka, M.; Anthony, R.M. Engineered Sialylation of Pathogenic Antibodies In Vivo Attenuates Autoimmune Disease. Cell 2018, 172, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of acute form of collagen antibody induced arthritis. Autoantibodies binding to well defined epitopes are transferred at day 0, followed by injection of lipopolysaccharide from E. coli 05:B55 as the secondary stimulus at day 3. Significant level of proteoglycan depletion was observed 72 h after antibody injection. Inflammation (red and swollenness) and, cartilage and bone erosions between arthritis and control mouse are shown. HE stained joint morphology of arthritis and control mice. Magnification, 10×. Pain (withdrawal threshold levels) started even before inflammation began and prolonged even after resolution of inflammation. Dotted arrows indicate the inserted figures.
Figure 1.
Schematic diagram of acute form of collagen antibody induced arthritis. Autoantibodies binding to well defined epitopes are transferred at day 0, followed by injection of lipopolysaccharide from E. coli 05:B55 as the secondary stimulus at day 3. Significant level of proteoglycan depletion was observed 72 h after antibody injection. Inflammation (red and swollenness) and, cartilage and bone erosions between arthritis and control mouse are shown. HE stained joint morphology of arthritis and control mice. Magnification, 10×. Pain (withdrawal threshold levels) started even before inflammation began and prolonged even after resolution of inflammation. Dotted arrows indicate the inserted figures.

Figure 2.
IgG dependent effector phase of arthritis. Binding of antibodies to epitopes present on the cartilage surface forms immune complexes leading to the activation of complement cascades and formation of anaphylatoxin, C5a, which attracts immune cells to the inflammatory foci. Antibodies also interact with FcγR bearing granulocytes, which secrete pro-inflammatory cytokines and proteases damaging cartilage and bone.
Figure 2.
IgG dependent effector phase of arthritis. Binding of antibodies to epitopes present on the cartilage surface forms immune complexes leading to the activation of complement cascades and formation of anaphylatoxin, C5a, which attracts immune cells to the inflammatory foci. Antibodies also interact with FcγR bearing granulocytes, which secrete pro-inflammatory cytokines and proteases damaging cartilage and bone.

Figure 3.
Bacterial enzymes as therapeutics for IgG dependent diseases. Streptococcus pyogenes secreted IdeS (A) cleaves IgG molecules at the hinge region and EndoS (B) cleaves N-linked carbohydrates specifically present on Fc region. Arthritis is ameliorated either after IgG-degrading enzyme (IdeS) or Endo-β-N-acetylglucosaminidase (EndoS) cleavage of pathogenic IgG (C). HE stained joint morphology of mouse with and without arthritis after treatment. Magnification, 10×.
Figure 3.
Bacterial enzymes as therapeutics for IgG dependent diseases. Streptococcus pyogenes secreted IdeS (A) cleaves IgG molecules at the hinge region and EndoS (B) cleaves N-linked carbohydrates specifically present on Fc region. Arthritis is ameliorated either after IgG-degrading enzyme (IdeS) or Endo-β-N-acetylglucosaminidase (EndoS) cleavage of pathogenic IgG (C). HE stained joint morphology of mouse with and without arthritis after treatment. Magnification, 10×.

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nandakumar, K.S. Targeting IgG in Arthritis: Disease Pathways and Therapeutic Avenues. Int. J. Mol. Sci. 2018, 19, 677. https://doi.org/10.3390/ijms19030677
AMA Style
Nandakumar KS. Targeting IgG in Arthritis: Disease Pathways and Therapeutic Avenues. International Journal of Molecular Sciences. 2018; 19(3):677. https://doi.org/10.3390/ijms19030677
Chicago/Turabian StyleNandakumar, Kutty Selva. 2018. "Targeting IgG in Arthritis: Disease Pathways and Therapeutic Avenues" International Journal of Molecular Sciences 19, no. 3: 677. https://doi.org/10.3390/ijms19030677
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.