
Autism Spectrum Disorders: Analysis of Mobile Elements at 7q11.23 Williams–Beuren Region by Comparative Genomics

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
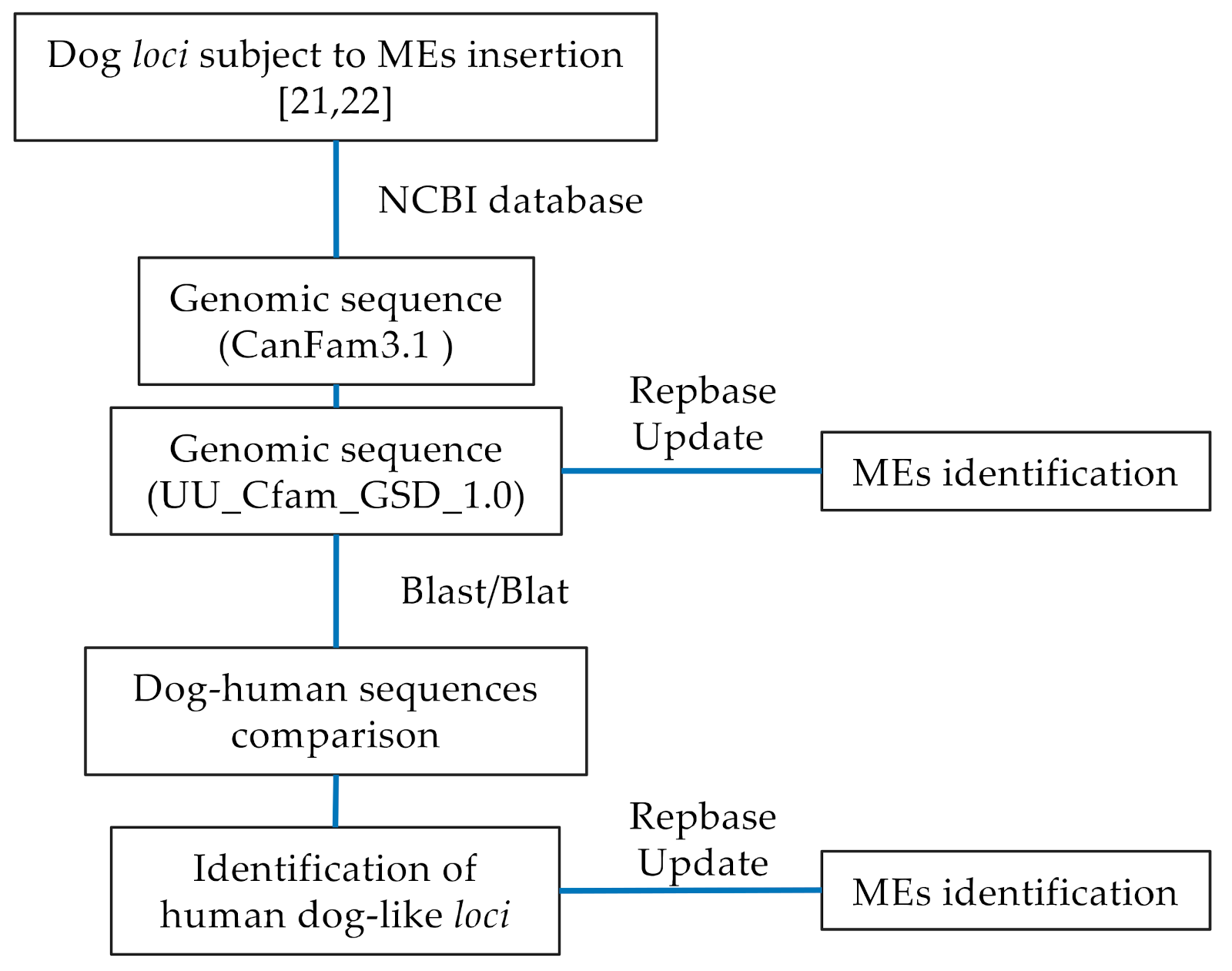
2.1. Comparative Genomic Analysis
2.2. MEs Detection in ASD Patients with ASD and Neurotypical Controls
2.2.1. Subjects
2.2.2. Diagnosis
2.2.3. Samples
2.2.4. Primer Design
2.2.5. PCR
2.2.6. Sanger Sequencing
2.2.7. Analysis of Genetic Variations
3. Results
3.1. Dog–Human Sequence Correspondence
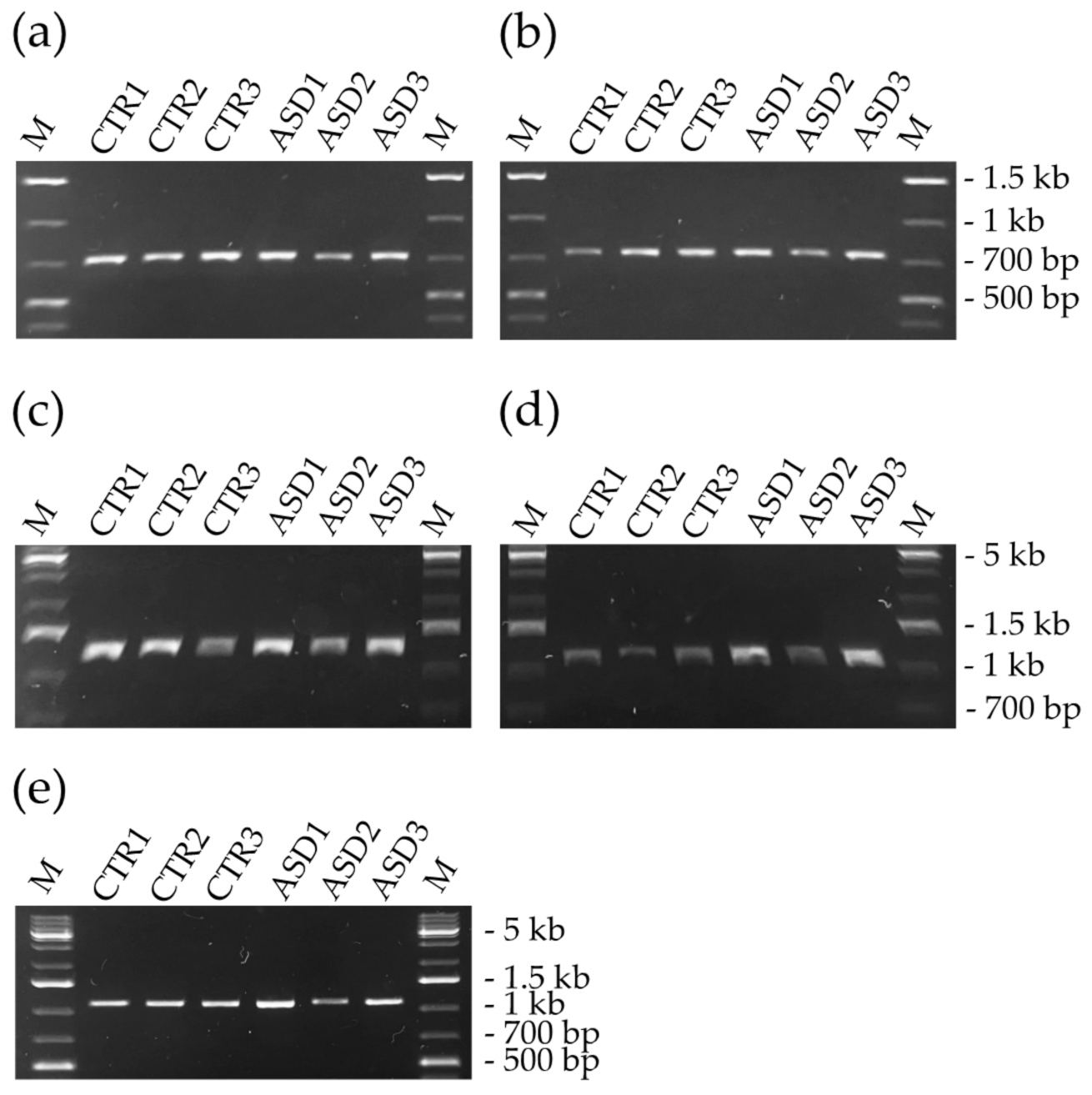
3.2. PCR Results
3.3. Sanger Sequencing Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adamo, A.; Atashpaz, S.; Germain, P.-L.; Zanella, M.; D’Agostino, G.; Albertin, V.; Chenoweth, J.; Micale, L.; Fusco, C.; Unger, C.; et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet. 2015, 47, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, M.W.M.; Verhoeven, E.W.M.; Geurts, H.M. Stop Making Noise! Auditory Sensitivity in Adults with an Autism Spectrum Disorder Diagnosis: Physiological Habituation and Subjective Detection Thresholds. J. Autism Dev. Disord. 2019, 49, 2116–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Manohar, H.; Kandasamy, P.; Chandrasekaran, V.; Rajkumar, R.P. Early Diagnosis and Intervention for Autism Spectrum Disorder: Need for Pediatrician-Child Psychiatrist Liaison. Indian J. Psychol. Med. 2019, 41, 87–90. [Google Scholar] [CrossRef]
- Mezzelani, A.; Landini, M.; Facchiano, F.; Raggi, M.E.; Villa, L.; Molteni, M.; De Santis, B.; Brera, C.; Caroli, A.M.; Milanesi, L.; et al. Environment, dysbiosis, immunity and sex-specific susceptibility: A translational hypothesis for regressive autism pathogenesis. Nutr. Neurosci. 2015, 18, 145–161. [Google Scholar] [CrossRef] [Green Version]
- Cheroni, C.; Caporale, N.; Testa, G. Autism spectrum disorder at the crossroad between genes and environment: Contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef]
- Troisi, J.; Autio, R.; Beopoulos, T.; Bravaccio, C.; Carraturo, F.; Corrivetti, G.; Cunningham, S.; Devane, S.; Fallin, D.; Fetissov, S.; et al. Genome, Environment, Microbiome and Metabolome in Autism (GEMMA) Study Design: Biomarkers Identification for Precision Treatment and Primary Prevention of Autism Spectrum Disorders by an Integrated Multi-Omics Systems Biology Approach. Brain Sci. 2020, 10, 743. [Google Scholar] [CrossRef]
- Berg, J.M.; Geschwind, D.H. Autism genetics: Searching for specificity and convergence. Genome Biol. 2012, 13, 247. [Google Scholar] [CrossRef] [Green Version]
- Rahnama, M.; Tehrani, H.A.; Mirzaie, M.; Ziaee, V. Identification of key genes and convergent pathways disrupted in autism spectrum disorder via comprehensive bioinformatic analysis. Inform. Med. Unlocked 2021, 24, 100589. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Williams, E.; Casanova, M.; Switala, A.; Li, H.; Qiu, M. Transposable elements occur more frequently in autism-risk genes: Implications for the role of genomic instability in autism. Transl. Neurosci. 2013, 4, 172–202. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Han, K.; Liang, P. Role of Transposable Elements in Gene Regulation in the Human Genome. Life 2021, 11, 118. [Google Scholar] [CrossRef]
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [Green Version]
- Lenhoff, H.M.; Wang, P.P.; Greenberg, F.; Bellugi, U. Williams syndrome and the brain. Sci. Am. 1997, 277, 68–73. [Google Scholar] [CrossRef]
- Schubert, C. The genomic basis of the Williams-Beuren syndrome. Cell. Mol. Life Sci. 2009, 66, 1178–1197. [Google Scholar] [CrossRef] [Green Version]
- Cuscó, I.; Corominas, R.; Bayés, M.; Flores, R.; Rivera-Brugués, N.; Campuzano, V.; Pérez-Jurado, L.A. Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res. 2008, 18, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.-J.; del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Pérez-Jurado, L.A.; et al. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, F.; Troglio, F.; Fagà, G.; Fancelli, D.; Shyti, R.; Trattaro, S.; Zanella, M.; D’Agostino, G.; Hughes, J.M.; Cera, M.R.; et al. High-throughput screening identifies histone deacetylase inhibitors that modulate GTF2I expression in 7q11.23 microduplication autism spectrum disorder patient-derived cortical neurons. Mol. Autism 2020, 11, 88. [Google Scholar] [CrossRef]
- MacLean, E.L.; Snyder-Mackler, N.; vonHoldt, B.M.; Serpell, J.A. Highly heritable and functionally relevant breed differences in dog behaviour. Proc. R. Soc. B Biol. Sci. 2019, 286, 20190716. [Google Scholar] [CrossRef] [Green Version]
- VonHoldt, B.M.; Shuldiner, E.; Koch, I.J.; Kartzinel, R.Y.; Hogan, A.; Brubaker, L.; Wanser, S.; Stahler, D.; Wynne, C.D.L.; Ostrander, E.A.; et al. Structural variants in genes associated with human Williams-Beuren syndrome underlie stereotypical hypersociability in domestic dogs. Sci. Adv. 2017, 3, e1700398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VonHoldt, B.M.; Ji, S.S.; Aardema, M.L.; Stahler, D.R.; Udell, M.A.R.; Sinsheimer, J.S. Activity of Genes with Functions in Human Williams-Beuren Syndrome Is Impacted by Mobile Element Insertions in the Gray Wolf Genome. Genome Biol. Evol. 2018, 10, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Tandon, D.; Ressler, K.; Petticord, D.; Papa, A.; Jiranek, J.; Wilkinson, R.; Kartzinel, R.Y.; Ostrander, E.A.; Burney, N.; Borden, C.; et al. Homozygosity for Mobile Element Insertions Associated with WBSCR17 Could Predict Success in Assistance Dog Training Programs. Genes 2019, 10, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Eyring, K.W.; Geschwind, D.H. Three decades of ASD genetics: Building a foundation for neurobiological understanding and treatment. Hum. Mol. Genet. 2021, 30, R236–R244. [Google Scholar] [CrossRef]
- Wang, C.; Wallerman, O.; Arendt, M.-L.; Sundström, E.; Karlsson, Å.; Nordin, J.; Mäkeläinen, S.; Pielberg, G.R.; Hanson, J.; Ohlsson, Å.; et al. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun. Biol. 2021, 4, 185. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S.L.; Luyster, R.J.; Guthrie, W. (Firm) Autism Diagnostic Observation Schedule: ADOS-2; Lord, C., Rutter, M., Eds.; Western Ps.: Torrance, CA, USA, 2012. [Google Scholar]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef]
- Werling, D.M.; Brand, H.; An, J.-Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Kleiber, M.L.; Whitney, J.; Maile, M.S.; Hong, O.; Chapman, T.R.; Tan, S.; Tandon, P.; et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 2018, 360, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Guffanti, G.; Gaudi, S.; Fallon, J.H.; Sobell, J.; Potkin, S.G.; Pato, C.; Macciardi, F. Transposable elements and psychiatric disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2014, 165, 201–216. [Google Scholar] [CrossRef]
- Guffanti, G.; Gaudi, S.; Klengel, T.; Fallon, J.H.; Mangalam, H.; Madduri, R.; Rodriguez, A.; DeCrescenzo, P.; Glovienka, E.; Sobell, J.; et al. LINE1 insertions as a genomic risk factor for schizophrenia: Preliminary evidence from an affected family. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2016, 171, 534–545. [Google Scholar] [CrossRef]
- Guffanti, G.; Bartlett, A.; DeCrescenzo, P.; Macciardi, F.; Hunter, R. Transposable Elements. Curr. Top. Behav. Neurosci. 2019, 42, 221–246. [Google Scholar] [CrossRef]
- Cisternas, F.A.; Vincent, J.B.; Scherer, S.W.; Ray, P.N. Cloning and characterization of human CADPS and CADPS2, new members of the Ca2+-dependent activator for secretion protein family. Genomics 2003, 81, 279–291. [Google Scholar] [CrossRef]
- Bonora, E.; Graziano, C.; Minopoli, F.; Bacchelli, E.; Magini, P.; Diquigiovanni, C.; Lomartire, S.; Bianco, F.; Vargiolu, M.; Parchi, P.; et al. Maternally inherited genetic variants of CADPS2 are present in autism spectrum disorders and intellectual disability patients. EMBO Mol. Med. 2014, 6, 795–809. [Google Scholar] [CrossRef]
- Tabuchi, K.; Südhof, T.C. Structure and evolution of neurexin genes: Insight into the mechanism of alternative splicing. Genomics 2002, 79, 849–859. [Google Scholar] [CrossRef]
- Südhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef] [Green Version]
- Trobiani, L.; Meringolo, M.; Diamanti, T.; Bourne, Y.; Marchot, P.; Martella, G.; Dini, L.; Pisani, A.; De Jaco, A.; Bonsi, P. The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2020, 119, 37–51. [Google Scholar] [CrossRef]
- Tromp, A.; Mowry, B.; Giacomotto, J. Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry 2021, 26, 747–760. [Google Scholar] [CrossRef]
- Khayat, W.; Hackett, A.; Shaw, M.; Ilie, A.; Dudding-Byth, T.; Kalscheuer, V.M.; Christie, L.; Corbett, M.A.; Juusola, J.; Friend, K.L.; et al. A recurrent missense variant in SLC9A7 causes nonsyndromic X-linked intellectual disability with alteration of Golgi acidification and aberrant glycosylation. Hum. Mol. Genet. 2019, 28, 598–614. [Google Scholar] [CrossRef] [Green Version]
- Schwede, M.; Garbett, K.; Mirnics, K.; Geschwind, D.H.; Morrow, E.M. Genes for endosomal NHE6 and NHE9 are misregulated in autism brains. Mol. Psychiatry 2014, 19, 277–279. [Google Scholar] [CrossRef]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; Yuce, T.; Kemper, T.; Bauman, M. Regional alterations in purkinje cell density in patients with autism. PLoS ONE 2014, 9, e81255. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; McLellan, A.S.; Ball, M.; Wynne, F.; O’Neill, C.; Mills, W.; Quinn, J.P.; Kleinjan, D.A.; Anney, R.J.; Carmody, R.J.; et al. Regulation of SPRY3 by X chromosome and PAR2-linked promoters in an autism susceptibility region. Hum. Mol. Genet. 2015, 24, 5126–5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Z.; Williams, J.M.; Kumari, R.; Baranov, P.V.; Moore, T. Opposite Expression Patterns of Spry3 and p75NTR in Cerebellar Vermis Suggest a Male-Specific Mechanism of Autism Pathogenesis. Front. Psychiatry 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Dog locus | Human Gene | Primer (Human) | Oligo Sequence (Human) | Product Size | Anealing Condition (°C) |
|---|---|---|---|---|---|
| Cfa6.6 | GALNT17 | Forward | ACATGGTCCTTCGCTAGAGAGA | 716 | 59.8 |
| Reverse | CCCCTTGGCCACCTAATCAA | ||||
| Cfa6.7 | NRXN3 | Forward | AGGTGCACATACTAAAACCAAATGA | 738 | 59 |
| Reverse | ACTGTTTTGTCCTCATGTCTTTTCA | ||||
| SLC9A7 | Forward | ACCCGGCCAACCTCTATTCA | 1089 | 60.5 | |
| Reverse | GCCACATATCAGACACCATCCT | ||||
| SPRY3 | Forward | CCCGGCCAGCAGTTTGTTAT | 1114 | 60.6 | |
| Reverse | TGACTTGCTCCAGGTGATAATCTG | ||||
| Cfa6.66 | GTF2IP14 | Forward | CATCCCCGAACAGCATTAACA | 1249 | 58.6 |
| Reverse | TGACCCATCATTACCAATCAGATTT |
| DOG (UU_Cfam_GSD_1.0) | Human (GRCh38.p13) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Gene | Chr | Amplicon Start | Amplicon End | TE Name | TE Class/Family | Chr | TE Start | TE End | Gene | Chr | Amplicon Start | Amplicon End | TE Name | TE Class/Family | Chr | TE Start | TE End |
| cfa6_6 | GALNT17 | 6 | 2,441,471 | 2,442,028 | MER5A | DNA/hAT-Charlie | 6 | 2,441,321 | 2,441,475 | GALNT17 | 7 | 71,198,462 | 71,199,177 | |||||
| (CA)n | Simple_repeat | 6 | 2,441,548 | 2,441,589 | ||||||||||||||
| SINEC2A1_CF | SINE/tRNA | 6 | 2,441,653 | 2,441,841 | ||||||||||||||
| cfa6_7 | GALNT17 | 6 | 2,466,130 | 2,466,636 | MER21C | LTR/ERVL | 6 | 2,465,886 | 2,466,144 | NRXN3 | 14 | 79,081,200 | 79,081,937 | AluSc8 | SINE/Alu | 14 | 79,081,335 | 79,081,634 |
| MER21C | LTR/ERVL | 6 | 2,466,179 | 2,466,334 | L2c | LINE/L2 | 14 | 79,081,635 | 79,081,755 | |||||||||
| SINEC2A1_CF | SINE/tRNA | 6 | 2,466,362 | 2,466,558 | SLC9A7 | X | 46,623,458 | 46,624,546 | MER21C | LTR/ERVL | X | 46,623,722 | 46,624,477 | |||||
| SPRY3 | X | 155,794,613 | 155,795,839 | MER21C | LTR/ERVL | X | 155,795,271 | 155,795,776 | ||||||||||
| cfa6_66 | GTF2I | 6 | 5,671,518 | 5,671,763 | GTF2IP14 | 7 | 65,095,549 | 65,096,634 | L1MC5a | LINE/L1 | 7 | 65,095,820 | 65,095,976 | |||||
| GTF2IP5 | 7 | 65,785,354 | 65,785,553 | |||||||||||||||
| Dog locus | Gene (Human) | Dog:Human Sequence Comparison Score | Dog:Human Sequence Identity (%) | ASD:CTR Consensus Identity (%) | SNP (N = novel) | Variation | MAF < 0.1 (true = 1; false = 0) | Chr | Start | End | Amplicons Sequenced (ASD) | ASD Carrying Variant | Amplicons Sequenced (CTR) | CTR Carrying Variant | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | n | % | n | n | % | |||||||||||
| Cfa6.6 | GALNT17 | 100 | 75.2 | 98.87 | rs1202647 | C/T | 0 | 7 | 71,198,716 | 71,198,716 | 10 | 5 | 50 | 10 | 4 | 40 |
| rs10260271 | A/G | 0 | 7 | 71,199,052 | 71,199,052 | 10 | 1 | 10 | 10 | 1 | 10 | |||||
| Cfa6.7 | NRNX3 | 48 | 78 | 95 | ||||||||||||
| Cfa6.7 | SPRY3 | 32 | 94.5 | 99.8 | N | G/A | X | 155,794,832 | 155,794,832 | 10 | 1 | 10 | 10 | 0 | 0 | |
| N | G/A | X | 155,794,906 | 155,794,906 | 10 | 1 | 10 | 10 | 0 | 0 | ||||||
| N | G/T | X | 155,795,335 | 155,795,335 | 10 | 1 | 10 | 10 | 0 | 0 | ||||||
| N | C/A | X | 155,795,522 | 155,795,522 | 10 | 0 | 0 | 10 | 1 | 10 | ||||||
| Cfa6.7 | SLC9A7 | 43 | 93 | 97.96 | rs1238052830 | T/A | 1 | X | 46,623,716 | 46,623,716 | 10 | 0 | 0 | 10 | 1 | 10 |
| N | A/G | X | 46,623,965 | 46,623,965 | 10 | 0 | 0 | 10 | 1 | 10 | ||||||
| N | T/C | X | 46,624,108 | 46,624,108 | 10 | 0 | 0 | 10 | 1 | 10 | ||||||
| N | C/T | X | 46,624,374 | 46,624,374 | 10 | 1 | 10 | 10 | 0 | 0 | ||||||
| Cfa6.66 | GTF2IP14 | 138 | 76.2 | 96.58 | rs1799101 | A/G | 0 | 7 | 65,095,563 | 65,095,563 | 7 | 7 | 100 | 8 | 8 | 100 |
| N | C/A | 7 | 65,095,595 | 65,095,595 | 7 | 0 | 0 | 7 | 1 | 14 | ||||||
| N | C/T | 7 | 65,095,612 | 65,095,612 | 8 | 4 | 50 | 9 | 5 | 56 | ||||||
| N | C/T | 7 | 65,095,616 | 65,095,616 | 9 | 2 | 22 | 9 | 2 | 22 | ||||||
| N | C/T | 7 | 65,095,634 | 65,095,634 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | C/T | 7 | 65,095,643 | 65,095,643 | 9 | 0 | 0 | 9 | 1 | 11 | ||||||
| N | G/A | 7 | 65,095,646 | 65,095,646 | 9 | 0 | 0 | 9 | 1 | 11 | ||||||
| N | G/A | 7 | 65,095,681 | 65,095,681 | 9 | 8 | 89 | 9 | 9 | 100 | ||||||
| N | C/A/T | 7 | 65,095,683 | 65,095,683 | 9 | 2 | 22 | 9 | 1 | 11 | ||||||
| N | T/A | 7 | 65,095,687 | 65,095,687 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | T/G | 7 | 65,095,689 | 65,095,689 | 9 | 0 | 0 | 9 | 1 | 11 | ||||||
| N | T/C | 7 | 65,095,690 | 65,095,690 | 9 | 0 | 0 | 9 | 1 | 11 | ||||||
| N | T/C | 7 | 65,095,698 | 65,095,698 | 9 | 1 | 11 | 9 | 1 | 11 | ||||||
| N | AG/CA | 7 | 65,095,709 | 65,095,710 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | insGGT | 7 | 65,095,712 | 65,095,713 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | T/C | 7 | 65,095,715 | 65,095,715 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | C/G | 7 | 65,095,718 | 65,095,718 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | T/C | 7 | 65,095,724 | 65,095,724 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | insCA | 7 | 65,095,728 | 65,095,729 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | A/G | 7 | 65,095,730 | 65,095,730 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | T/A | 7 | 65,095,742 | 65,095,742 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | C/T | 7 | 65,095,746 | 65,095,746 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | C/T | 7 | 65,095,753 | 65,095,753 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | insCA | 7 | 65,095,754 | 65,095,755 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | A/G | 7 | 65,095,755 | 65,095,755 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | C/T/A | 7 | 65,095,765 | 65,095,765 | 9 | 2 | 22 | 9 | 1 | 11 | ||||||
| N | C/T | 7 | 65,095,778 | 65,095,778 | 9 | 1 | 11 | 9 | 1 | 11 | ||||||
| N | C/A | 7 | 65,095,779 | 65,095,779 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | C/T | 7 | 65,095,833 | 65,095,833 | 9 | 1 | 11 | 9 | 0 | 0 | ||||||
| N | G/A | 7 | 65,096,185 | 65,096,185 | 9 | 5 | 56 | 9 | 4 | 44 | ||||||
| rs10262238 | C/T | 0 | 7 | 65,096,197 | 65,096,197 | 9 | 4 | 44 | 9 | 5 | 56 | |||||
| N | A/C | 7 | 65,096,281 | 65,096,281 | 10 | 1 | 10 | 10 | 1 | 10 | ||||||
| N | T/A | 7 | 65,096,298 | 65,096,298 | 10 | 10 | 100 | 10 | 10 | 100 | ||||||
| rs71562961 | A/G | 0 | 7 | 65,096,467 | 65,096,467 | 10 | 2 | 20 | 10 | 1 | 10 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cupaioli, F.A.; Fallerini, C.; Mencarelli, M.A.; Perticaroli, V.; Filippini, V.; Mari, F.; Renieri, A.; Mezzelani, A. Autism Spectrum Disorders: Analysis of Mobile Elements at 7q11.23 Williams–Beuren Region by Comparative Genomics. Genes 2021, 12, 1605. https://doi.org/10.3390/genes12101605
Cupaioli FA, Fallerini C, Mencarelli MA, Perticaroli V, Filippini V, Mari F, Renieri A, Mezzelani A. Autism Spectrum Disorders: Analysis of Mobile Elements at 7q11.23 Williams–Beuren Region by Comparative Genomics. Genes. 2021; 12(10):1605. https://doi.org/10.3390/genes12101605
Chicago/Turabian StyleCupaioli, Francesca Anna, Chiara Fallerini, Maria Antonietta Mencarelli, Valentina Perticaroli, Virginia Filippini, Francesca Mari, Alessandra Renieri, and Alessandra Mezzelani. 2021. "Autism Spectrum Disorders: Analysis of Mobile Elements at 7q11.23 Williams–Beuren Region by Comparative Genomics" Genes 12, no. 10: 1605. https://doi.org/10.3390/genes12101605