
Prader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene

Abstract
:1. Introduction
2. The MAGEL2 Gene in Prader-Willi Syndrome and Schaaf-Yang Syndrome

{kind=link}
{kind=link}
| Patient | Mutation Type | Age | Sex | Height | Height Z-Score | Weight | Weight Z-Score | BMI | BMI Z-Score |
|---|---|---|---|---|---|---|---|---|---|
| Schaaf et al. subject 1 | Truncating point mutation | 13 yo | M | 156 cm | −0.03 | 54.2 kg | 0.82 | 22.3 | 1.13 |
| Schaaf et al. subject 2 | Truncating point mutation | 7 yr 6 mo | M | 134 cm | 1.65 | 68.3 kg | 3 | 38 | 2.99 |
| Schaaf et al. subject 3 | Truncating point mutation | 5 yo | M | 105 cm | −0.91 | 19.6 kg | 0.42 | 17.8 | 1.56 |
| Schaaf et al. subject 4 | Truncating point mutation | 19 yo 5 mo | M | 148 cm | −3 | 47.9 kg | −2.84 | 21.9 | −0.29 |
| Soden et al. subject 382 | Truncating point mutation | 11 yo | F | 140 cm | −0.55 | 60.8 kg | 2.02 | 31 | 2.35 |
| Soden et al. subject 383 | Truncating point mutation | 8 yo | F | 107 cm | −3 | 16.7 kg | −3 | 14.6 | −0.77 |
| Kanber et al. patient 1 | Gene deletion | 7 yo | F | 140 cm | 3 | 41 kg | 2.6 | 20.9 | 1.92 |
| Buiting et al. patient 1 | Gene deletion | 3 yo | M | 102.5 cm | 1.86 | 17.3 kg | 1.59 | 16.5 | 0.37 |
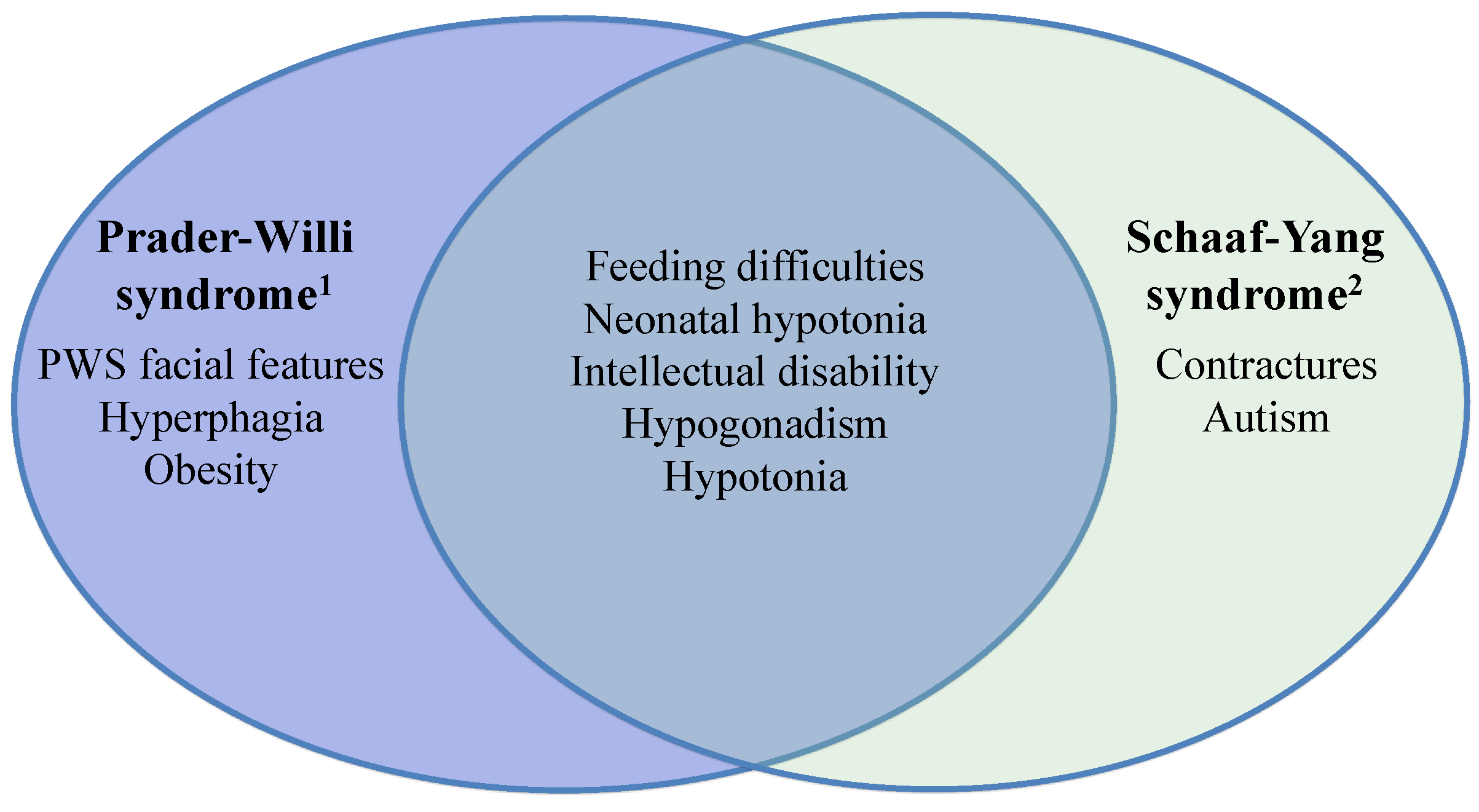
2.1. Schaaf-Yang Syndrome vs. Prader-Willi Syndrome
2.2. Animal Models of Magel2 Loss-of-Function

| Wevrick et al. [19,20,21,22] | Muscatelli et al. [17,18] |
|---|---|
| Postnatal lethality (10%) | Neonatal lethality (50%) |
| Decreased pre-wean weight gain | Decreased oxytocin quantity |
| Increased post-wean adiposity | Decreased suckling activity |
| Decreased lean mass | Decreased sociability (male mice) |
| Decreased locomotor activity | Decreased learning and memory (male mice) |
| Altered appreciation of novelty | |
| Circadian dysfunction | |
| Progressive infertility | |
| Altered olfaction (>24 weeks of age) |
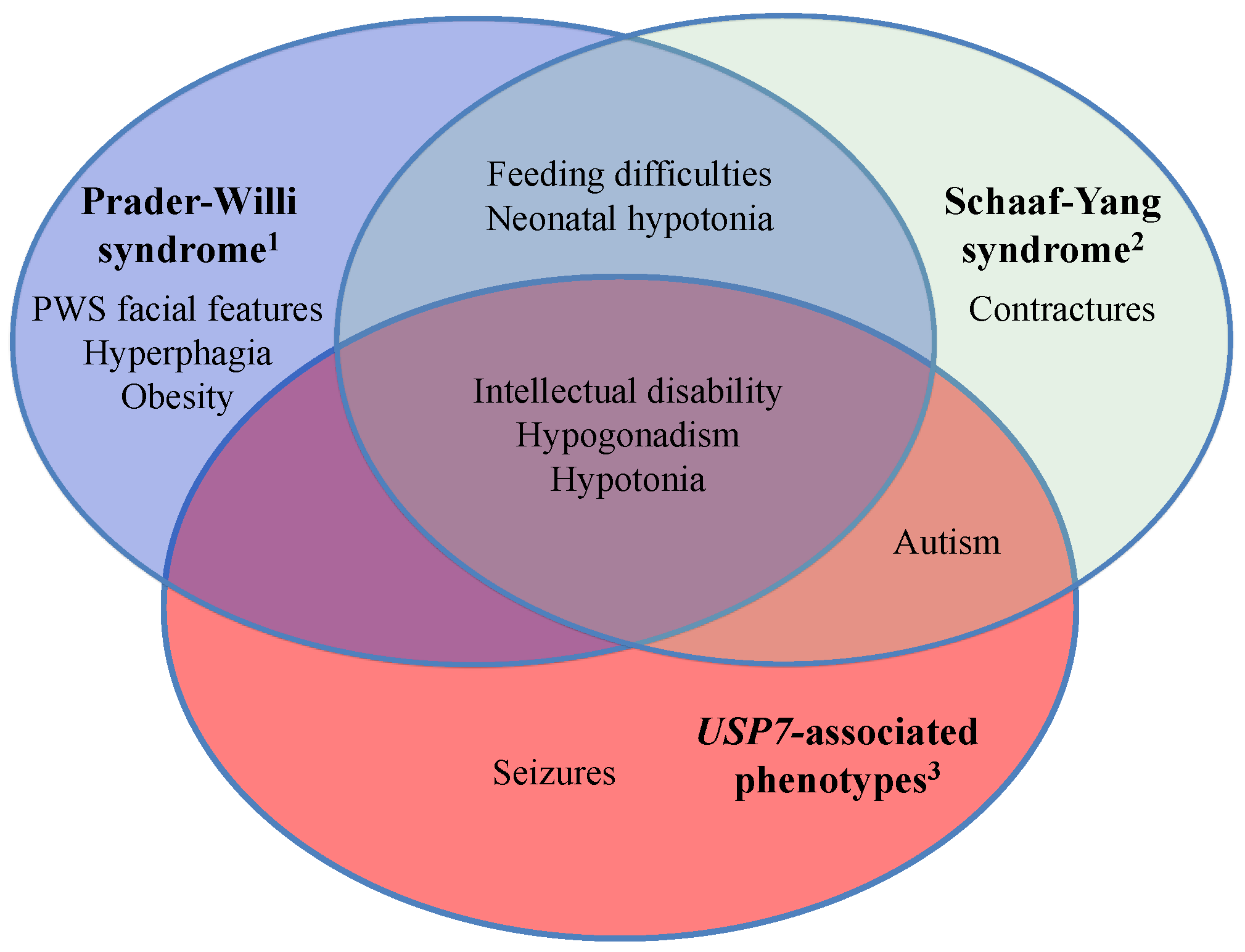
2.3. USP7 Haploinsufficiency and Its Related Clinical Phenotypes

3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar] [PubMed]
- Dykens, E.M.; Lee, E.; Roof, E. Prader-Willi syndrome and autism spectrum disorders: An evolving story. J. Neurodev. Disord. 2011, 3, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.J.; Einfeld, S.L. Cognitive and behavioural aspects of Prader-Willi syndrome. Curr. Opin. Psychiatry 2015, 28, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Roof, E. Behavior in Prader-Willi syndrome: Relationship to genetic subtypes and age. J. Child Psychol. Psychiatry 2008, 9, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in Prader-Willi syndrome: A systematic review. Am. J. Med. Genet. A 2015, 167, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K. Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef] [PubMed]
- De Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Duker, A.L.; Ballif, B.C.; Bawle, E.V.; Person, R.E.; Mahadevan, S.; Alliman, S.; Thompson, R.; Traylor, R.; Bejjani, B.A.; Shaffer, L.G.; et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 2010, 18, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Kanber, D.; Giltay, J.; Wieczorek, D.; Zogel, C.; Hochstenbach, R.; Caliebe, A.; Kuechler, A.; Horsthemke, B.; Buiting, K. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; di Donato, N.; Beygo, J.; Bens, S.; von der Hagen, M.; Hackmann, K.; Horsthemke, B. Clinical phenotypes of MAGEL2 mutations and deletions. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, V.; Muscatelli, F. Natural breaking of the maternal silence at the mouse and human imprinted Prader-Willi locus: A whisper with functional consequences. Rare Dis. 2013, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Soden, S.E.; Saunders, C.J.; Willig, L.K.; Farrow, E.G.; Smith, L.D.; Petrikin, J.E.; Lepichon, J.; Miller, N.A.; Thiffault, I.; Dinwiddie, D.L.; et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 2014, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mejlachowicz, D.; Nolent, F.; Maluenda, J.; Ranjatoelina-Randrianaivo, H.; Giuliano, F.; Gut, I.; Sternberg, D.; Laquerrière, A.; Melki, J. Truncating mutations of MAGEL2, a gene within the Prader-Willi locus, are responsible for severe arthrogryposis. Am. J. Hum. Genet. 2015, 97, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Schaller, F.; Watrin, F.; Sturny, R.; Massacrier, A.; Szepetowski, P.; Muscatelli, F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted MAGEL2 gene. Hum. Mol. Genet. 2010, 19, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Schaller, F.; Bauer, S.; Villard, C.; Matarazzo, V.; Riet, F.; Guillon, G.; Lafitte, D.; Desarmenien, M.G.; Tauber, M.; et al. An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for MAGEL2, a gene involved in Prader-Willi syndrome and autism. Biol. Psychiatry 2014, 78, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.M.; Stewart, C.L.; Wevrick, R. Inactivation of the mouse MAGEL2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 2007, 16, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.E.; Kwolek, E.M.; Bischof, J.M.; van Eede, M.; Henkelman, R.M.; Wevrick, R. Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene MAGEL2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.E.; Wevrick, R. Loss of MAGEL2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PLoS ONE 2009, 4, e4291. [Google Scholar] [CrossRef] [PubMed]
- Tennese, A.A.; Wevrick, R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in MAGEL2-null mice. Endocrinology 2011, 152, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.-H.; Fountain, M.D.; Tacer, K.F.; Xia, F.; Bi, W.; Kang, S.-H.L.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol. Cell 2015, 59, 956–969. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fountain, M.D.; Schaaf, C.P. Prader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene. Diseases 2016, 4, 2. https://doi.org/10.3390/diseases4010002
Fountain MD, Schaaf CP. Prader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene. Diseases. 2016; 4(1):2. https://doi.org/10.3390/diseases4010002
Chicago/Turabian StyleFountain, Michael D., and Christian P. Schaaf. 2016. "Prader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene" Diseases 4, no. 1: 2. https://doi.org/10.3390/diseases4010002