
- Division of Medical Oncology, Department of Internal Medicine, The James Comprehensive Cancer Center, The Ohio State University Wexner Medical Center, Columbus, OH, USA
Gynecologic malignancies annually account for over 91,000 new cancer cases and approximately 28,000 deaths in the United States. Although there have been advancements in cytotoxic chemotherapies, there has not been significant improvement in overall survival in these patients. While targeted therapies have shown some benefit in many solid tumors, further development of these agents is needed for the treatment of gynecologic malignancies. Poly(ADP-ribose) polymerase (PARP) catalyzes the polyADP-ribosylation of proteins involved in DNA repair. Inhibitors of PARP were originally developed for cancers with homologous recombination deficiencies, such as those harboring mutations in BRCA1 or BRCA2 genes. However, pre-clinical research and clinical trials have suggested that the activity of PARP inhibitors is not limited to those with BRCA mutations. PARP inhibitors may have activity in cancers deficient in other DNA repair genes, signaling pathways that mitigate DNA repair, or in combination with DNA-damaging agents independent of DNA repair dysfunction. Currently there are seven different PARP inhibitors in clinical development for cancer. While there has been promising clinical activity for some of these agents, there are still significant unanswered questions regarding their use. Going forward, specific questions that must be answered include timing of therapy, use in combination with cytotoxic agents or as single-agent maintenance therapy, and whether there is a predictive biomarker that can be used with PARP inhibition. Even with large strides in the treatment of many gynecologic malignancies in recent years, it is imperative that we develop newer agents and methods to identify patients that may benefit from these compounds. The focus of this review will be on pre-clinical data, current clinical trials, and the future of PARP inhibitors in the treatment of ovarian, endometrial, and cervical cancer.
Introduction
Gynecologic malignancies annually account for over 91,000 new cancer cases and approximately 28,000 deaths in the United States (1). Effective screening for cervical cancer is available in many parts of the world, but there is no effective screening for endometrial or ovarian cancer. Many women with ovarian cancer, therefore, present with advanced stage disease for which cure is rare. Endometrial cancer is more commonly diagnosed early on, as patients are often symptomatic with postmenopausal bleeding. While there have been advancements in the development and administration of cytotoxic chemotherapies, there has not been significant improvement in overall survival in these patients. It is imperative that novel and effective treatment strategies are developed. Although targeted therapies have shown occasional benefit in some solid tumors, these agents have been largely ineffective for the treatment of gynecologic malignancies.
One area of recent interest in targeted therapies for many cancers has been the development of poly(ADP-ribose) polymerase (PARP) inhibitors. PARP catalyzes the polyADP-ribosylation of proteins involved in DNA repair. Inhibitors of PARP were shown to be highly selective for cancer cells that harbor homologous recombination (HR) deficiencies, such as those harboring mutations in BRCA1 or BRCA2 genes (2). PARP inhibitors cause an increase in single strand breaks (SSBs) in DNA that, if left unrepaired, will lead to double strand breaks (DSBs) when encountered by replication forks (3, 4). In the laboratory, HR-deficient cells are unable to maintain genomic integrity in the presence of a large number of DNA DSBs and are, therefore, exquisitely sensitive to PARP inhibition. This synthetic lethal interaction between PARP and BRCA has been proposed as a potential explanation for the sensitivity of BRCA mutation cell lines to PARP inhibition. Pre-clinical research and clinical trials, however, have suggested that the activity of PARP inhibitors is not limited to those with BRCA mutations. PARP inhibitors may demonstrate synthetic lethality in cancers deficient in other proteins that mitigate DNA repair (5). McCabe et al. examined the effects of PARP inhibition on various cell lines deficient in RAD51, Fanconi anemia complementation group (FANC), and Nijmegen breakage syndrome 1 (NBS1), amongst other proteins involved in HR, and found that mutations of these individual proteins induced sensitivity to PARP (6). These findings suggest that the notion of synthetic lethality may be more broadly applied to cancers with an impaired HR pathway, not just those with BRCA mutations. This concept is frequently referred to as “BRCAness” or “BRCA-like” (7). The inhibition of SSB repair by PARP inhibition may also be sufficient to enhance the anti-cancer activity in combination with DNA-damaging agents independent of dysfunction in DNA repair pathways (8).
The combination of phosphatase and tensin homolog (PTEN)-deficient cells and PARP inhibition is another area of potential synergistic activity. PTEN encodes for a phosphatase that negatively regulates the phosphatidylinositide 3-kinase (PI3K)/AKT/mTOR pathway, which is important for cell proliferation and survival (9, 10) and also plays a poorly understood role in the expression of the DNA repair protein RAD51 and in the functionality of HR. Both in vitro and in vivo studies have demonstrated sensitivity of PTEN-deficient cells to PARP inhibitors (11–13). Thus, PARP inhibition may benefit patients with malignancies in which there is decreased PTEN expression, such as endometrial cancer, glioblastoma, malignant melanoma, prostate, breast, lung, and colorectal cancers (11).
Currently, there are multiple PARP inhibitors in clinical development for cancer. While there has been promising clinical activity for some of these agents, there are still significant unanswered questions regarding their use. Going forward, specific questions that must be answered include: timing of therapy, use in combination with cytotoxic agents or as a single-agent, maintenance therapy, and the existence of predictive biomarker(s) that can be used with PARP inhibition. Even with large strides in the treatment of many gynecologic malignancies in recent years, it is imperative that we develop newer agents and methods to identify patients that may benefit from these compounds.
Poly(ADP-Ribose) Polymerase
Base excision repair (BER) is one of multiple critical pathways that maintain genome integrity in all cells, specifically in the recognition and repair of SSBs (14, 15). PARP is a family of 17 proteins that play an important role in DNA repair pathways. The most well studied member of the family, PARP1, is critical in the BER pathway for DNA SSBs. It detects and binds single strand DNA damage sites through its zinc finger domains, next attaching poly(ADP) ribose (PAR) moieties on itself and other proteins that have been recruited to the damage site (Figure 1). If there is excessive DNA damage, such as is seen with ischemia, PARP1 becomes hyperactivated. This heightened activity results in high levels of PAR and the depletion of nicotinamide adenine dinucleotide (NAD+) and adenosine triphosphate (ATP) (16), and ultimately, cell death termed parthanatos (17). PARP is also involved in the repair of DSBs (18) and the recruitment of additional repair proteins like ataxia telangiectasia-mutated (ATM) and mitotic recombination 11 (MRE11), both of which are integral to the HR process (19, 20).
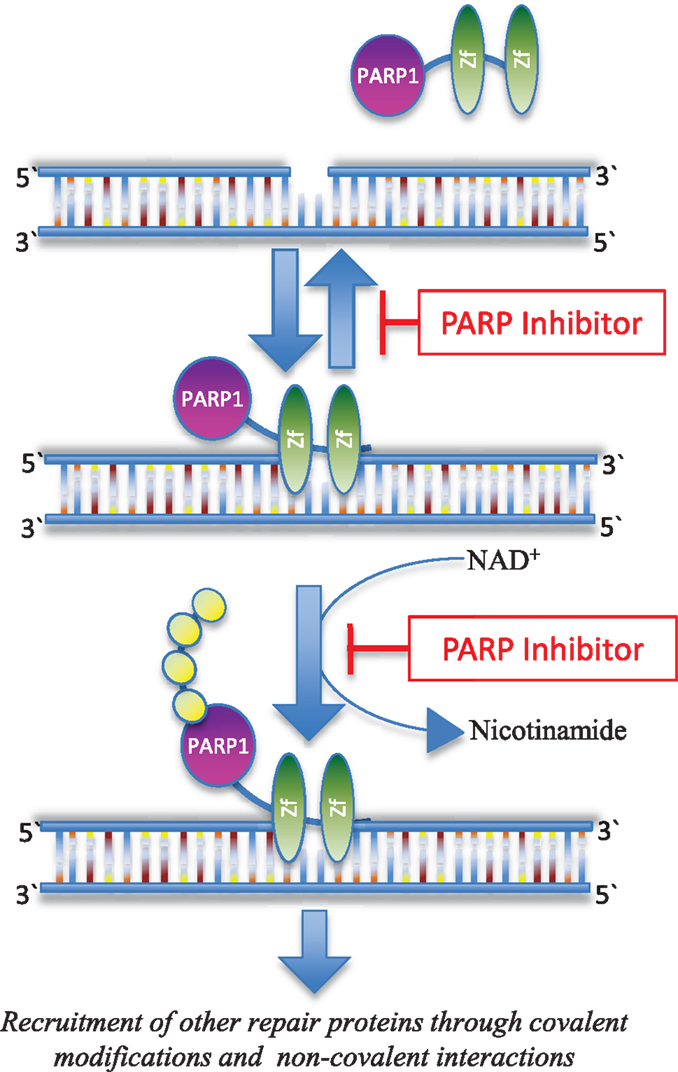
Figure 1. Inhibition of PARP function. PARP1 recognizes and binds to sites of DNA damage through its Zn-finger domains (Zf). PARP inhibitors can trap PARP1 on damaged DNA in a complex that is possibly more toxic than unrepaired single strand DNA breaks (28). PARP inhibitors also block the enzymatic activity of the enzyme thereby inhibiting poly(ADP-ribosyl)ation, which in turn blocks recruitment of downstream repair proteins (114).
PARP1 was first reported in 1963 (21), but its anti-cancer utility was not fully realized until 1980. At that time, Durkacz et al. demonstrated that early-generation PARP inhibitors not only hindered DNA repair, but also enhanced the cytotoxic effects of DNA methylating agents in murine leukemia (22). Kupper et al. demonstrated the enhancement of the cytotoxic effects of gamma-irradiation after reduction of active PARP through overexpression of a dominant negative mutant of PARP that recognizes and binds damaged DNA, but does not possess the catalytic activity of the enzyme (23). More recently, PARP moved into the spotlight with the discovery that PARP inhibition in both cancer cell lines (2, 24) and human tumors (25) lacking BRCA1 or BRCA2 is selectively cytotoxic compared to non-mutation containing tumors. One rationale for this efficacy is a principle termed synthetic lethality, a condition by which deletion or inactivation of only one of two genes (either BRCA or PARP) would not cause cell death, but deletion or inactivation of two genes in combination (both BRCA and PARP) is lethal. If PARP1 and PARP2 are inhibited, SSBs typically repaired by BER remain unresolved and when encountered by a replication fork, lead to the accumulation of DSBs (26). BRCA1- or BRCA2-deficient cells lack the ability to effectively complete HR and repair DNA DSBs. This double hit by impairment of both BRCA and PARP functionality ultimately results in genomic instability and cell death. Conferring a potential therapeutic benefit, cell death appears to be limited to homozygous target tissues (i.e., tumor), since most BRCA patients carry only one copy of the wild-type BRCA gene and there is no apparent effect on cells heterozygous for BRCA mutations (2). These observations have been exploited in the treatment of cancers associated with BRCA mutations, such as hereditary breast and ovarian cancer (HBOC), and even endometrial cancers (27).
Recently, Murai et al. suggest that the action of PARP inhibition is not only a function of how well the inhibitors disrupt the enzymatic activity, but that certain inhibitors also trap PARP1 on damaged DNA, thereby blocking repair (28). Interestingly, these studies showed that the potency in trapping PARP1 varied among agents, independent of their catalytic inhibitory properties. Clearly, additional investigation is warranted to better understand the intricacies inherent to PARP inhibition pathway and ultimately, advance drug development.
Hereditary Breast and Ovarian Cancer and BRCA
Hereditary breast and ovarian cancer is typically characterized by the onset of breast cancer at a young age, a strong family history of both breast and ovarian cancer, as well as an autosomal dominant inheritance pattern. Fallopian tube and primary peritoneal cancers also fall into this hereditary spectrum and are included under the ovarian cancer designation. An increased chance of bilateral cancers (e.g., both breasts), the development of both breast and ovarian cancer, and/or an increased incidence of other cancers (pancreas, prostate, etc.) may also be seen in this syndrome. In ovarian cancer, 10% of patients have a genetic predisposition. However, in those patients with a family history of ovarian cancer, the rate of BRCA1 mutations is 80 and 15% for BRCA2 mutations (29). More recently with the use of a massively parallel sequencing approach, Walsh et al. identified that closer to 24% of serous ovarian cancer patients have a germline DNA repair defect, over 30% of these were in patients without a family history of breast or ovarian cancer (30). The use of this broader assay is a promising method for detecting germline mutations with greater sensitivity and at decreased cost. Approximately 5–10% of all breast cancers and up to 25–40% of breast cancers in young patients (<35 years old) are hereditary. An estimated 3–8% of all breast cases and 30–40% of familial cases are likely caused by BRCA1 and BRCA2 mutations.
Individuals with a BRCA mutation have an increased risk of developing ovarian cancer up to 63% by some estimates, and breast cancer by up to 87% (31). Patients with BRCA1 breast tumors tend to have a higher histologic grade, medullary histopathology, and are more likely than sporadic (non-BRCA mutant) tumors to be estrogen receptor negative, progesterone-receptor negative, and HER2/neu overexpression negative or “triple negative” (TNBC) (32). Ovarian cancers associated with BRCA1 mutation are more often serous adenocarcinomas (90%) compared to women without this mutation (50%) (33–36). Although largely derived from retrospective or indirect data, most studies have not identified a significant survival difference between individuals with BRCA mutation-associated breast cancer versus controls (37–44). However, patients with high-grade serous ovarian carcinoma associated with a BRCA mutation tend to have a better prognosis than sporadic cases (45, 46). This improved prognosis may be related to BRCA-mutated cells’ impaired DNA repair mechanism, lending these lesions greater sensitivity to cytotoxic chemotherapy, especially with platinum-based agents (47, 48). Based on the high selective lethality of BRCA-mutated cancer cells to PARP inhibitors, multiple studies have been undertaken to establish efficacy in gynecologic malignancies.
The Role of PARP Inhibitors in Ovarian Cancer
Although it ranks as the ninth most common cancer among women, excluding non-melanoma skin cancers, ovarian cancer is the fifth most deadly cancer in females and accounts for more deaths than any other cancer of the female reproductive tract in the United States (1). Since the symptoms of disease are typically non-specific, ovarian cancer is often detected in advanced stages when the chance of cure is low. Given its insidious nature and the lethality of the disease, novel therapies are needed to improve overall survival in ovarian cancer patients.
In BRCA mutation-associated ovarian cancers, multiple investigations have been completed or are presently underway to establish the clinical activity of PARP inhibition in these mutational carriers. Sixty patients with refractory solid tumors were enrolled in a phase I trial of the PARP inhibitor olaparib (KU-0059436/AZD2281); the study was enriched for patients with BRCA mutations (25). In addition to establishing the maximum tolerated dose (MTD) of olaparib at 400 mg bid and observing only minimal adverse effects (primarily fatigue and gastrointestinal), it was noted that only BRCA mutation carriers had a significant objective tumor response. Out of 19 patients, 9 had a partial response (PR) (47%) and remarkably, 8 of which were ovarian cancer patients. Twelve of these patients (63%) had either radiological or tumor-marker responses or stable disease for ≥4 months. In an expanded cohort of the same trial, 50 patients with BRCA1/2 mutation-associated ovarian, primary peritoneal, and fallopian tube cancers were found to have a clinical benefit rate of 46%, including 40% that experienced a Response Evaluation Criteria in Solid Tumors (RECIST) radiologic or CA125 response (49). The median duration of response was 28 weeks. Another key finding was the overall clinical benefit rate was correlated with platinum sensitivity. Platinum-resistant and refractory patients had a 46 and 23% respective benefit rate versus 69% in the platinum-sensitive population (P = 0.038). The study also reported statistically significant associations between the overall platinum-free interval and antitumor response, as well as between platinum sensitivity and the maximum percentage change from radiologic baseline tumor size and from baseline CA125 after olaparib treatment.
In a phase 2 international, multicenter fashion, two sequential cohorts of women with confirmed BRCA1 or 2 mutations and recurrent disease were given either olaparib at 400 mg twice daily (n = 33) or 100 mg twice daily (n = 24) (50). The primary efficacy endpoint was objective response rate (ORR). In the 400 mg twice-daily cohort, ORR was 11 of 33 patients (33%; 95% CI 20–51); in the 100 mg twice daily cohort, ORR was 3 of 24 patients (13%; 95% CI 4–31). The most common toxicities experienced included nausea, anemia, and fatigue and were mild in the majority of cases. This phase 2 study provided positive proof of concept for the efficacy and tolerability of olaparib in advanced BRCA-mutated ovarian cancer.
Stemming from these initial reports, Kaye et al. designed a phase II, open-label, randomized, international study to assess the safety and efficacy of different doses (200 or 400 mg) of olaparib given twice daily versus intravenous liposomal doxorubicin given monthly in patients with BRCA-related ovarian cancer who had failed prior platinum-based chemotherapy (51). A statistically significant higher combined RECIST and CA125 rate of response for olaparib 400 mg twice daily compared to liposomal doxorubicin was noted. It did not find a significant difference in progression free survival (PFS) between the groups, with a reported median PFS of 7.1 months for liposomal doxorubicin, 6.5 months for the 200 mg olaparib cohort, and 8.8 months for the 400 mg olaparib cohort. There were roughly twice as many ≥grade 3 toxicities seen with liposomal doxorubicin compared to the PARP inhibitor. While this study did not show a statistically significant improvement in PFS between olaparib and liposomal doxorubicin, there was a much greater PFS with liposomal doxorubicin (7.1 months) than had been reported in historical data. Gordon et al. demonstrated PFS was only 4 months for liposomal doxorubicin compared to topotecan in a phase III randomized study of recurrent ovarian cancer (52). A recently reported phase III trial by Colombo et al. also demonstrated a similar PFS (3.7 months) for liposomal doxorubicin (53). Although the ability to draw comparisons between studies is limited, Kaye et al. reported PFS with liposomal doxorubicin is still within the 95% CI of historical controls, which suggests that this difference may simply reflect random variation within the population (54).
In addition to their use in BRCA mutation-associated ovarian cancer, PARP inhibitors are also being investigated in non-mutation carrier (or BRCA wild-type) ovarian cancers. Using PARP inhibitors in such a scenario is based on the idea that there is a HR DNA repair defect, but no germline BRCA1/2 mutation in up to 50% of ovarian cancers (7, 11, 46, 55). Several studies have exploited this concept. Gelmon et al. conducted a phase II trial with high-grade serous/undifferentiated ovarian cancer with unknown BRCA status or BRCA-negative disease (56) and an additional reference group with known germline BRCA mutations. Patients were treated with olaparib 400 mg twice daily. The ORR in BRCA-mutants (n = 17) was 41% (95% CI 22–64) with median PFS of 221 days (95% CI 106–383), while BRCA mutation negative patients had an ORR of 24% (n = 46; 95% CI 14–38) and PFS of 192 days (95% CI 109–267). In a post hoc exploratory analysis, the ORR in patients with platinum-sensitive ovarian cancer was 50% (10 of 20) in the BRCA-negative cohort and 60% (3 of 5) in the BRCA-mutant cohort. In platinum-resistant ovarian cancers, 33 and 4% of patients with BRCA mutation positive and BRCA-mutant negative status respectively had responses. Observed toxicities were similar to those described in previous studies. This trial’s findings were noteworthy, as they solidified the clinical utility of PARP inhibition in sporadic ovarian cancer. Further, these results suggest that platinum sensitivity may be used as a surrogate marker for HR deficiency. Results of a phase I study of niraparib (MK4827), an oral PARP inhibitor shown to induce selective lethality in HR repair deficient tumors with BRCA loss or non-BRCA HR defects (57), was given to a small cohort of patients enriched for BRCA-deficient and sporadic cancers associated with HR repair defects (58). Thirty-nine patients were treated at 7 successive dose levels; 11 of these patients were BRCA mutation carriers. Although results are only available in abstract form, the study reported that three patients with serous ovarian cancer had prolonged RECIST PR (one sporadic platinum-sensitive, two BRCA-deficient ovarian cancers). Disease stabilization was observed for >44 weeks in the sporadic serous ovarian cancer patient and for >16 weeks in the two patients with BRCA-deficient disease. In another phase II study with the PARP inhibitor rucaparib (AG-014699/PF-0136738), 41 patients with either breast (17) or ovarian (24) cancer and known BRCA deficiencies were given rucaparib as monotherapy and followed for ORR (59). Preliminary findings included a clinical benefit rate of 32%, but an ORR of 5% (2/38). However, 26% (10/38) achieved stable disease for ≥4 months and three patients remained on study for >54 weeks. The final results from these two ongoing studies are anxiously awaited.
Another larger, randomized, double-blind, placebo-controlled, phase II trial evaluated maintenance treatment with olaparib in patients with platinum-sensitive, relapsed, high-grade serous ovarian cancer (60). Included patients had received ≥2 platinum-based regimens and were required to have had a partial or complete response to their most recent platinum-based therapy. Two-hundred and sixty five patients were randomized to receive olaparib at 400 mg twice daily or placebo (136 olaparib arm, 129 placebo). BRCA mutational status was similar between the two groups. PFS was significantly longer in the olaparib arm than placebo (8.4 versus 4.8 months); however, there was no difference in overall survival at the first interim analysis. Interestingly, subgroup analysis revealed that regardless of BRCA mutational status, the olaparib cohort had a decreased risk for progression. Toxicities were overall mild in the olaparib group; most adverse events were grade 1 or 2 and typically included nausea, fatigue, vomiting, and anemia. These findings again support the argument that platinum sensitivity is a useful clinical marker for olaparib sensitivity. Further, this investigation recapitulates the role of PARP inhibitors in the ovarian cancer population, regardless of BRCA mutational status, and underscores the need for development of relevant biomarkers that predict HR deficiency in the setting of BRCA mutations or no known genetic abnormalities. Fortunately, there are multiple ongoing trials investigating the relationship between PARP inhibition and ovarian cancer that will hopefully clarify some of these uncertainties (Table 1).
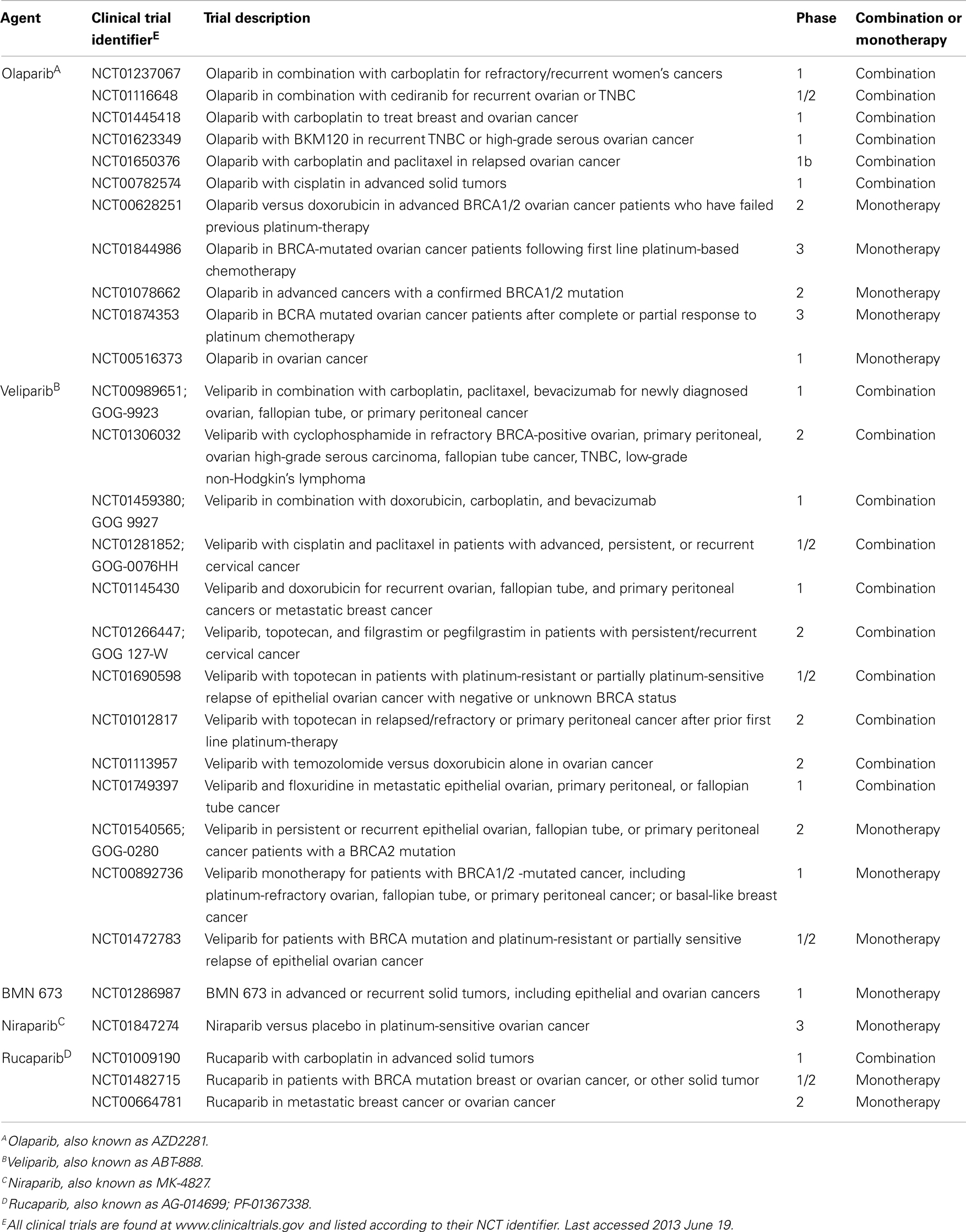
Table 1. Active clinical trials investigating PARP inhibitors in gynecologic malignancies.
PARP Inhibitors in Endometrial Cancer
Endometrial cancer is the fourth most common cancer in women and the most commonly diagnosed gynecologic malignancy. An estimated 90% of the cases are sporadic and 10% have a genetic origin. Endometrioid adenocarcinoma and serous carcinoma are the most prevalent histological types, while endometrial clear cell and mucinous carcinomas only account for approximately 5% of all cases (61). Since many patients are symptomatic early in their disease course, the majority of endometrial cancers (approximately 75%) are detected in the initial stages when the disease remains confined to the uterus (61). However, a significant amount of women still experience advanced disease, for which systemic treatment options are limited, toxicities high, and responses often short-lived (62, 63). There is a pressing need for targeted therapies that will yield a greater efficacy and be better tolerated.
A variety of different molecular defects linked to the development of endometrial cancer are described. In endometrioid endometrial carcinoma (EEC), also known as type I endometrial cancer, microsatellite instability (MSI) and mutations in the PTEN, K-ras, PIK3CA, and β-catenin genes are reported (64). As previously discussed, PTEN is a tumor suppressor gene that is involved in DNA repair mechanisms, as well as in the inhibition of the PI3K/AKT/mTOR pathway; PTEN-deficient cells are sensitive to PARP inhibitors (11–13). Rare syndromes collectively known as the PTEN hamartoma tumor syndromes (PHTS) are linked to germline mutations in PTEN (65, 66). Outside of PHTS, PTEN is altered in up to 83% of endometrioid carcinomas versus only 10% in serous and clear cell cancers (67–71). Dedes et al. demonstrated that PTEN-deficient EEC cells had a greater sensitivity to PARP inhibition than wild-type EEC PTEN cell lines (12). Given the heightened prevalence of PTEN deficiency in EEC superimposed on these laboratory studies demonstrating sensitivity to PARP inhibition, clinical studies are now in progress. A case report describing a 58-year-old female with metastatic endometrioid endometrial adenocarcinoma who had previously demonstrated exquisite sensitivity to platinum-containing regimens, was given olaparib as part of a phase I trial (72, 73). Prior to trial participation, brain metastases were found. However, after 10 weeks on trial, the patient had a significant reduction in the size of the brain metastases without other intervention and also reported improvement in tumor-related symptoms. Unfortunately, the patient had objective disease progression after 8 months on olaparib therapy. Her tumor was biopsied and verified to be negative for BRCA mutation, but positive for loss of PTEN. Although only an isolated report, this case study coupled with compelling pre-clinical data, provides a strong rationale for larger clinical trials. A phase 2, randomized, placebo-controlled trial comparing olaparib versus best supportive care or progesterone in advanced endometrial cancer was planned, but unfortunately, was unable to be opened. In addition to EEC, serous endometrial cancers appear to have a similar genetic background to serous ovarian carcinoma, including hallmarks of deficiency in DNA repair as well as frequent mutations in TP53, PIK3CA, K-RAS, and ERBB2 (74). These tumors may prove to be another rational target for PARP inhibition.
PARP Inhibitors in Cervical Cancer
As the third most common cancer worldwide, cervical cancer has an annual incidence of 530,000 cases, with 250,000 deaths expected (75). It is the second leading cause of death in women from the ages of 20–39 (76). Fortunately, the incidence of this cancer in most developed countries has decreased by 70% over the past 50 years due to improved screening methods with cervical cytology (77). More recently, HPV vaccination has aided in the detection and subsequent prevention of high-risk HPV subtypes, which are the culprit for most cervical cancers (78–82). For advanced disease, chemotherapy remains the standard of care. Similar to the experience in endometrial cancer, such therapy typically does not yield durable responses or cure (83).
The use of PARP inhibitors in cervical cancer has only recently been explored in the pre-clinical arena. Along with non-small cell lung cancer, mesothelioma, and ovarian cancer cell lines, Michels et al. created cervical cancer (HeLa) cell lines resistant to cisplatin (84). Upon further study, these lines were found to have high levels of PAR and PARP1, with PARP1 constitutively hyperactivated. Exposure of the cells to pharmacologic PARP inhibition resulted in cell death. Hence, this work hints at another role for PARP inhibition, in the treatment of cisplatin-resistant cervical cancers. Interestingly, this group also observed that elevated levels of PAR identified in PARP1-overexpressing tumor cells and xenografts predicted response to PARP inhibition in vitro and in vivo more accurately than PARP1 expression itself, suggesting PAR may be a reasonable biomarker of response to PARP inhibitor therapy in cervical cancer. A phase I trial is presently enrolling patients with cervical cancer along with other gynecological malignancies to investigate the combination of olaparib with carboplatin in refractory or recurrent disease (NCT01237067; see Table 1). Another phase 1/2 trial is investigating the use of veliparib with cisplatin and paclitaxel in advanced, persistent, or recurrent cervical cancer (NCT01281852; Table 1). Additional pre-clinical and clinical investigation will hopefully reveal even more promising applications for PARP inhibition in cervical cancer.
Future Directions
Poly(ADP-ribose) polymerase inhibitors are an exciting new class of agents that have already demonstrated promising pre-clinical and clinical activity in a variety of malignancies. Nevertheless, the full potential of PARP inhibition in cancer has not yet been realized. In addition to single-agent use, PARP inhibitors have been studied in combination with a number of different chemotherapies, anti-angiogenic agents, as well as with ionizing radiation. Other areas of active investigation include the development of markers that will predict clinical benefit from PARP inhibition, as well as the identification of resistance mechanisms to PARP inhibitor therapy.
Chemotherapies known to induce DNA strand breaks, especially SSBs, are of particular interest for combination studies. In the case of methylating agents, activation of BER elicits therapy resistance (85). A large body of pre-clinical in vivo and in vitro studies demonstrates the addition of a PARP inhibitor may sensitize cells to DNA-damaging agents and further delay the development of treatment resistance (8, 85–93). These studies were conducted with a wide variety of chemotherapeutic agents, including topoisomerase I inhibitors, platinum agents, as well as DNA alkylating agents. Human trials combining PARP inhibitors and chemotherapy agents for sporadic and BRCA-associated gynecologic malignancies are underway, but few have reached maturity (NCT01445418, NCT01237067; see Table 1). Promising data has come from Oza et al., who conducted a multicenter phase II study that compared the efficacy of olaparib plus paclitaxel/carboplatin followed by olaparib maintenance therapy versus paclitaxel/carboplatin alone with no further therapy in patients with platinum-sensitive recurrent serous ovarian cancer (94). Importantly, the BRCA status was unknown for the majority of the patients. In arm A, patients received six, 21-day cycles of olaparib (200 mg twice daily) with paclitaxel (175 mg/m2 IV, day 1) and carboplatin (AUC 4 IV, day 1), followed by olaparib maintenance therapy at a dose of 400 mg twice daily in a continuous fashion versus in arm B, the standard dose of carboplatin (AUC 6 IV, day 1) and paclitaxel (175 mg/m2 IV, day 1) without the PARP inhibitor. Patients receiving olaparib had a significant improvement in PFS versus chemotherapy alone. OS data was felt to be immature, but preliminarily showed similar results between the two arms (64 versus 58%). In the combination phase, both arms had generally similar toxicity profiles, with nausea, fatigue, and alopecia the most common adverse events experienced. During the maintenance phase (olaparib monotherapy versus no further therapy), side effects were consistent with the known monotherapy side effect profile of PARP inhibitors. In a smaller phase I dose escalation trial, olaparib was added to carboplatin in BRCA1/2 mutational carriers with breast or ovarian cancer (95). Therapy was administered in a 3 × 3 dose escalation fashion: oral olaparib at 100 or 200 mg every 12 h [dose level (DL) 1/2] with IV carboplatin AUC 3 on day 8 then every 21 days; DL6–9 gave olaparib days 1–7 at 200 then 400 mg every 12 h, with carboplatin AUC 3 on day 2 then escalation to AUC 5 (no DL3–5). From the preliminary results, bone marrow suppression was the observed dose limiting toxicity. Of the 23 evaluable ovarian cancer patients, PR was seen in 8/23, disease stabilization occurred in 11/23. Overall, the ovarian cancer cohort had a clinical benefit of 83%. Clearly, the results of these studies are intriguing; data from similar combination trials is eagerly anticipated.
In addition to chemotherapeutic agents, PARP inhibitors are also being combined with anti-angiogenic agents. The rationale behind this combination is based on the observation that vascular endothelial growth factor receptor (VEGFR) inhibition may lead to increased DNA damage through downregulation of DNA repair proteins, including ERCC1 and XRCC1 (96, 97). Stemming from pre-clinical data supporting the relationship between PARP inhibition and the VEGF pathway (98–100), several phase I studies are presently underway. The phase 1 study of ABT-888 (veliparib) in combination with carboplatin, paclitaxel, and bevacizumab as first-line treatment for stage II-IV ovarian cancer is actively enrolling patients (NCT00989651; Table 1). Another phase I trial of olaparib in combination with cediranib, a VEGFR inhibitor, is also open to recurrent ovarian or TNBC patients (NCT01116648; Table 1). Trial investigators are exploring the toxicities and recommended phase 2 dosing of the dual therapy. From a preliminary report, myelosuppression was dose limiting at the highest dose level (cediranib 30 mg daily/olaparib 400 mg twice daily) (101). Although unconfirmed, the study also notes a 56% response rate in enrolled ovarian cancer patients. These results are encouraging; additional efficacy data will be forthcoming (Table 1).
Due to PARP’s ability to inhibit multiple processes related to DNA repair, combining PARP inhibition with ionizing radiation is a logical combination. Pre-clinical studies confirm that PARP inhibition acts to sensitize malignant cells to radiation (88, 102). Several laboratories have also shown that PARP 1 knockout mice have an enhanced sensitivity to gamma-radiation (103, 104). In mouse colon cancer xenografts, veliparib coupled with irradiation resulted in prolonged survival from 23 to 36 days, and in one mouse, a complete response (8). At the present time, there are no active clinical trials investigating the combination of radiation therapy with PARP inhibition in gynecologic malignancies. However, there are active trials investigating this dual therapy in other diseases like breast cancer (NCT01477489) (105) and glioblastoma multiforme (NCT00687765) (106). Enrollment of gynecologic malignancy patients into similar trials is important since radiation plays a significant role in the treatment of cervical and endometrial cancer.
As evidenced by the discussed clinical data, many patients benefit from PARP inhibitor therapy, though the degree of response varies and sometimes there is no observed clinical benefit. A predictive marker that not only evaluates the drug’s pharmacodynamic effects, but can also identify who might benefit from therapy may help guide treatment decisions. Several attempts have been made to meet this objective. Duan et al. described a triple stain immunofluorescence assay looking at FANCD2, DAPI, and Ki67 as a means for measuring the functional competency of the Fanconi anemia pathway in proliferating cells in formalin fixed tumor tissue from patient biopsies across multiple tumor types (5). This stain is now being tested in a prospective fashion to select patients for a phase 1 clinical trial using veliparib alone or in combination with mitomycin-C (NCT01017640). The use of massively parallel sequencing analysis (e.g., BROCA) in a prospectively designed trial should also be investigated as this may capture a larger percentage of patients likely to be sensitive to PARP inhibition compared to relying on BRCA1/2 mutational analysis alone (30). Mukhopadhyay et al. developed a method of measuring HR function by quantifying RAD51 foci via immunofluorescence-based assays of ascitic fluid (107). They subsequently correlated in vitro cytotoxicity of the PARP inhibitor rucaparib with the HR status from these culture results. They correlated their in vitro results to patients whom were treated with platinum-based chemotherapy; tumor progression and OS were prospectively compared between HR-competent versus HR-deficient patients (108). Interestingly, patients who were HR-deficient, as established by assay analysis, had lower rates of tumor progression at 6 months and a higher median survival. From these results, the authors suggest that the RAD51 assay successfully identified those patients with HR deficiency and hence, may better predict which patients will have the best response to PARP inhibition. In addition to ascitic fluid, collection of peripheral blood mononuclear cells (PBMCs) as a surrogate tissue to monitor drug actions may be preferable to tumor biopsy collection, as it is less invasive and multiple samples may be longitudinally obtained. In order to better characterize the pharmacodynamic profile of the PARP inhibitor ABT-888, Ji et al. developed an immunoassay for measuring PAR incorporation in both tumor biopsies and PBMCs (109). In this study, considerable inter-individual and inter-sample heterogeneity in PAR levels was observed. Given these findings, it is not surprising that the trial comparing cyclophosphamide with veliparib presented a 50% reduction in PAR levels in 90% of patient PBMCs and 80% reduction in tumor biopsies across all dose levels (110). A larger phase II follow up study with this combination is ongoing (NCT01306032; Table 1). Though limited conclusions may be drawn from this experience, one must consider the possibility that PAR levels did not correlate well with actual PARP inhibitor activity (111). Ongoing genomic microarray analysis of patients involved in trials using olaparib may give useful insight into genetic signatures that may predict response. Regardless, these results underscore the need to identify a validated method of quantifying PARP inhibitor activity that corresponds to actual clinical outcome.
As with the majority of anti-cancer agents, tumors may develop acquired resistance to PARP inhibitor therapy. There are several proposed mechanisms of resistance, and likely many more that have not yet been described. One potential means is the restoration of HR secondary to a gain of function mutation in the BRCA2 allele via elimination of the c.6174delT mutation (112). Resistance secondary to up regulation of the ABCB1a/b gene that encodes for a P-glycoprotein efflux pump is also described with long-term use of the PARP inhibitor olaparib. Reversal of resistance occurred with co-administration of a P-glycoprotein inhibitor (113). These are just two examples of methods of resistance and certainly the success of PARP inhibitor therapy in the future will rely on further analysis of resistance patterns and subsequent therapy modifications.
Conclusion
Gynecologic malignancies represent a significant challenge in women’s health. When discovered in advanced stages, few successful therapeutic interventions are available to patients. Therefore, the development of novel agents like PARP inhibitors is essential. Already recognized as a promising agent in the treatment of BRCA-related malignancy, initial phase I and II studies confirm the activity of PARP inhibitors in ovarian, endometrial, and cervical cancers. As we learn more about these targeted agents through ongoing trials, it will be important to identify which population of patients may benefit the most from PARP inhibitor therapy and in what manner, as monotherapy or in combination. Whether it is in the neoadjuvant, adjuvant, or maintenance setting, the timing of therapy that will procure the greatest clinical benefit is also unknown. Clearly, PARP inhibitors are an exciting new class of targeted agents for the treatment of ovarian, endometrial, and cervical cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ATP, adenosine triphosphate; ATM, ataxia telangiectasia-mutated; BER, base excision repair; DL, dose level; DSB, double strand breaks; EEC, endometrioid endometrial carcinoma; FANC, Fanconi anemia complementation group; HBOC, hereditary breast and ovarian cancer; HR, homologous recombination; MRE11, mitotic recombination 11; MSI, microsatellite instability; MTD, maximum tolerated dose; NAD+, nicotinamide adenine dinucleotide; NSB1, Nijmegen breakage syndrome; ORR, objective response rate; PAR, poly(ADP) ribose; PARP, poly(ADP-ribose) polymerase; PFS, progression free survival; PI3K, phosphatidylinositide 3-kinase; PHTS, PTEN hamartoma tumor syndromes; PMBCs, peripheral blood mononuclear cells; RECIST, response evaluation criteria in solid tumors; SSBs, single strand breaks; TNBC, triple negative breast cancer; VEGFR, vascular endothelial growth factor receptor.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi:10.3322/caac.21166
2. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi:10.1038/nature03445
3. Boulton S, Kyle S, Durkacz BW. Interactive effects of inhibitors of poly(ADP-ribose) polymerase and DNA-dependent protein kinase on cellular responses to DNA damage. Carcinogenesis (1999) 20(2):199–203. doi:10.1093/carcin/20.2.199
4. Saffhill R, Ockey CH. Strand breaks arising from the repair of the 5-bromodeoxyuridine-substituted template and methyl methanesulphonate-induced lesions can explain the formation of sister chromatid exchanges. Chromosoma (1985) 92(3):218–24. doi:10.1007/BF00348697
5. Duan W, Gao L, Zhao W, Leon M, Sadee W, Webb A, et al. Assessment of FANCD2 nuclear foci formation in paraffin-embedded tumors: a potential patient-enrichment strategy for treatment with DNA interstrand crosslinking agents. Transl Res (2013) 161(3):156–64. doi:10.1016/j.trsl.2012.09.003
6. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res (2006) 66(16):8109–15. doi:10.1158/0008-5472.CAN-06-0140
7. Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer (2004) 4(10):814–9. doi:10.1038/nrc1457
8. Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res (2007) 13(9):2728–37. doi:10.1158/1078-0432.CCR-06-3039
9. Stambolic V, Suzuki A, dela Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell (1998) 95(1):29–39. doi:10.1016/S0092-8674(00)81780-8
10. Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A (1998) 95(26):15587–91. doi:10.1073/pnas.95.26.15587
11. Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med (2009) 1(6-7):315–22. doi:10.1002/emmm.200900041
12. Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med (2010) 2(53):53ra75. doi:10.1126/scitranslmed.3001538
13. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res (2010) 70(13):5457–64. doi:10.1158/0008-5472.CAN-09-4295
14. Dianov GL, Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res (2013) 41(6):3483–90. doi:10.1093/nar/gkt076
15. Odell ID, Wallace SS, Pederson DS. Rules of engagement for base excision repair in chromatin. J Cell Physiol (2013) 228(2):258–66. doi:10.1002/jcp.24134
16. Carson DA, Carrera CJ, Wasson DB, Yamanaka H. Programmed cell death and adenine deoxynucleotide metabolism in human lymphocytes. Adv Enzyme Regul (1988) 27:395–404. doi:10.1016/0065-2571(88)90028-3
17. David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (2009) 14:1116–28. doi:10.2741/3297
18. Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res (2006) 34(21):6170–82. doi:10.1093/nar/gkl840
19. Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem (2008) 283(2):1197–208. doi:10.1074/jbc.M706734200
20. Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet (2003) 40(10):721–8. doi:10.1136/jmg.40.10.721
21. Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun (1963) 11:39–43. doi:10.1016/0006-291X(63)90024-X
22. Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature (1980) 283(5747):593–6. doi:10.1038/283593a0
23. Kupper JH, Muller M, Jacobson MK, Tatsumi-Miyajima J, Coyle DL, Jacobson EL, et al. trans-dominant inhibition of poly(ADP-ribosyl)ation sensitizes cells against gamma-irradiation and N-methyl-N’-nitro-N-nitrosoguanidine but does not limit DNA replication of a polyomavirus replicon. Mol Cell Biol (1995) 15(6):3154–63.
24. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434(7035):913–7. doi:10.1038/nature03443
25. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361(2):123–34. doi:10.1056/NEJMoa0900212
26. Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol (2005) 25(16):7158–69. doi:10.1128/MCB.25.16.7158-7169.2005
27. Pennington KP, Walsh T, Lee M, Pennil C, Novetsky AP, Agnew KJ, et al. BRCA1, TP53, and CHEK2 germline mutations in uterine serous carcinoma. Cancer (2013) 119(2):332–8. doi:10.1002/cncr.27720
28. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res (2012) 72(21):5588–99. doi:10.1158/0008-5472.CAN-12-2753
29. Lux MP, Fasching PA, Beckmann MW. Hereditary breast and ovarian cancer: review and future perspectives. J Mol Med (Berl) (2006) 84(1):16–28. doi:10.1007/s00109-005-0696-7
30. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A (2011) 108(44):18032–7. doi:10.1073/pnas.1115052108
31. Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science (1994) 265(5181):2088–90. doi:10.1126/science.8091231
32. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol (2008) 26(15):2568–81. doi:10.1200/JCO.2007.13.1748
33. Berchuck A, Heron KA, Carney ME, Lancaster JM, Fraser EG, Vinson VL, et al. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res (1998) 4(10):2433–7.
34. Aida H, Takakuwa K, Nagata H, Tsuneki I, Takano M, Tsuji S, et al. Clinical features of ovarian cancer in Japanese women with germ-line mutations of BRCA1. Clin Cancer Res (1998) 4(1):235–40.
35. Lu KH, Cramer DW, Muto MG, Li EY, Niloff J, Mok SC. A population-based study of BRCA1 and BRCA2 mutations in Jewish women with epithelial ovarian cancer. Obstet Gynecol (1999) 93(1):34–7. doi:10.1016/S0029-7844(98)00362-7
36. Rubin SC, Benjamin I, Behbakht K, Takahashi H, Morgan MA, LiVolsi VA, et al. Clinical and pathological features of ovarian cancer in women with germ-line mutations of BRCA1. N Engl J Med (1996) 335(19):1413–6. doi:10.1056/NEJM199611073351901
37. Gaffney DK, Brohet RM, Lewis CM, Holden JA, Buys SS, Neuhausen SL, et al. Response to radiation therapy and prognosis in breast cancer patients with BRCA1 and BRCA2 mutations. Radiother Oncol (1998) 47(2):129–36. doi:10.1016/S0167-8140(98)00023-1
38. Johannsson OT, Ranstam J, Borg A, Olsson H. Survival of BRCA1 breast and ovarian cancer patients: a population-based study from southern Sweden. J Clin Oncol (1998) 16(2):397–404.
39. Verhoog LC, Brekelmans CT, Seynaeve C, Dahmen G, van Geel AN, Bartels CC, et al. Survival in hereditary breast cancer associated with germline mutations of BRCA2. J Clin Oncol (1999) 17(11):3396–402.
40. Verhoog LC, Brekelmans CT, Seynaeve C, van den Bosch LM, Dahmen G, van Geel AN, et al. Survival and tumour characteristics of breast-cancer patients with germline mutations of BRCA1. Lancet (1998) 351(9099):316–21. doi:10.1016/S0140-6736(97)07065-7
41. Brekelmans CT, Tilanus-Linthorst MM, Seynaeve C, vd Ouweland A, Menke-Pluymers MB, Bartels CC, et al. Tumour characteristics, survival and prognostic factors of hereditary breast cancer from BRCA2-, BRCA1- and non-BRCA1/2 families as compared to sporadic breast cancer cases. Eur J Cancer (2007) 43(5):867–76. doi:10.1016/j.ejca.2006.12.009
42. Rennert G, Bisland-Naggan S, Barnett-Griness O, Bar-Joseph N, Zhang S, Rennert HS, et al. Clinical outcomes of breast cancer in carriers of BRCA1 and BRCA2 mutations. N Engl J Med (2007) 357(2):115–23. doi:10.1056/NEJMoa070608
43. Kriege M, Seynaeve C, Meijers-Heijboer H, Collee JM, Menke-Pluymers MB, Bartels CC, et al. Sensitivity to first-line chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol (2009) 27(23):3764–71. doi:10.1200/JCO.2008.19.9067
44. Budroni M, Cesaraccio R, Coviello V, Sechi O, Pirino D, Cossu A, et al. Role of BRCA2 mutation status on overall survival among breast cancer patients from Sardinia. BMC Cancer (2009) 9:62. doi:10.1186/1471-2407-9-62
45. Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol (2008) 26(32):5284–93. doi:10.1200/JCO.2008.18.1107
46. Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol (2008) 26(34):5530–6. doi:10.1200/JCO.2008.16.1703
47. Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer (2003) 97(9):2187–95. doi:10.1002/cncr.11310
48. Cannistra SA. Cancer of the ovary. N Engl J Med (2004) 351(24):2519–29. doi:10.1056/NEJMra041842
49. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol (2010) 28(15):2512–9. doi:10.1200/JCO.2009.26.9589
50. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet (2010) 376(9737):245–51. doi:10.1016/S0140-6736(10)60893-8
51. Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol (2012) 30(4):372–9. doi:10.1200/JCO.2011.36.9215
52. Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol (2001) 19(14):3312–22.
53. Colombo N, Kutarska E, Dimopoulos M, Bae DS, Rzepka-Gorska I, Bidzinski M, et al. Randomized, open-label, phase III study comparing patupilone (EPO906) with pegylated liposomal doxorubicin in platinum-refractory or -resistant patients with recurrent epithelial ovarian, primary fallopian tube, or primary peritoneal cancer. J Clin Oncol (2012) 30(31):3841–7. doi:10.1200/JCO.2011.38.8082
54. Konstantinopoulos PA, Cannistra SA. Comparing poly (ADP-ribose) polymerase inhibitors with standard chemotherapy in BRCA-mutated, recurrent ovarian cancer: lessons learned from a negative trial. J Clin Oncol (2012) 30(4):347–50. doi:10.1200/JCO.2011.40.1489
55. Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol (2008) 26(19):3259–67. doi:10.1200/JCO.2007.11.3902
56. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol (2011) 12(9):852–61. doi:10.1016/S1470-2045(11)70214-5
57. Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem (2009) 52(22):7170–85. doi:10.1021/jm901188v
58. Sandhu SK, Wenham RM, Wilding G, McFadden M, Sun L, Toniatti C, et al. First-in-human trial of a poly(ADP-ribose) polymerase (PARP) inhibitor MK-4827 in advanced cancer patients (pts) with antitumor activity in BRCA-deficient and sporadic ovarian cancers (ASCO Meeting Abstracts). J Clin Oncol (2010) 28(15):3001. [Accessed 2013 Jun 16]. Available at: http://meeting.ascopubs.org/cgi/content/abstract/28/15_suppl/3001
59. Drew Y, Ledermann JA, Jones A, Hall G, Jayson GC, Highley M, et al. Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2-mutated, advanced ovarian and/or locally advanced or metastatic breast cancer (ASCO Meeting Abstracts). J Clin Oncol (2011) 29(15):3104. [Accessed 2013 Jun 16]. Available at: http://meeting.ascopubs.org/cgi/content/short/29/15_suppl/3104?rss=1
60. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med (2012) 366(15):1382–92. doi:10.1056/NEJMoa1105535
61. Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet (2005) 366(9484):491–505. doi:10.1016/S0140-6736(05)67063-8
62. Dellinger TH, Monk BJ. Systemic therapy for recurrent endometrial cancer: a review of North American trials. Expert Rev Anticancer Ther (2009) 9(7):905–16. doi:10.1586/era.09.54
63. Fleming GF. Systemic chemotherapy for uterine carcinoma: metastatic and adjuvant. J Clin Oncol (2007) 25(20):2983–90. doi:10.1200/JCO.2007.10.8431
64. Llaurado M, Ruiz A, Majem B, Ertekin T, Colas E, Pedrola N, et al. Molecular bases of endometrial cancer: new roles for new actors in the diagnosis and the therapy of the disease. Mol Cell Endocrinol (2012) 358(2):244–55. doi:10.1016/j.mce.2011.10.003
65. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet (1997) 16(1):64–7. doi:10.1038/ng0597-64
66. Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med (2009) 11(10):687–94. doi:10.1097/GIM.0b013e3181ac9aea
67. Bansal N, Yendluri V, Wenham RM. The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control (2009) 16(1):8–13.
68. Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst (2000) 92(11):924–30. doi:10.1093/jnci/92.11.924
69. Temkin SM, Fleming G. Current treatment of metastatic endometrial cancer. Cancer Control (2009) 16(1):38–45.
70. Ioffe YJ, Chiappinelli KB, Mutch DG, Zighelboim I, Goodfellow PJ. Phosphatase and tensin homolog (PTEN) pseudogene expression in endometrial cancer: a conserved regulatory mechanism important in tumorigenesis? Gynecol Oncol (2012) 124(2):340–6. doi:10.1016/j.ygyno.2011.10.011
71. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Cancer Genome Atlas Research Network, et al. Integrated genomic characterization of endometrial carcinoma. Nature (2013) 497(7447):67–73. doi:10.1038/nature12113
72. Molife LR, Forster MD, Krebs M, Pwint T, Middleton MR, Kaye SB, et al. A phase I study to determine the comparative bioavailability of two different oral formulations of the PARP inhibitor, olaparib (AZD2281), in patients with advanced solid tumors (ASCO Meeting Abstracts). J Clin Oncol (2010) 28(15):2599. [Accessed 2013 Jun 7]. Available at: http://meeting.ascopubs.org/cgi/content/abstract/28/15_suppl/2599
73. Forster MD, Dedes KJ, Sandhu S, Frentzas S, Kristeleit R, Ashworth A, et al. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol (2011) 8(5):302–6. doi:10.1038/nrclinonc.2011.42
74. Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A (2013) 110(8):2916–21. doi:10.1073/pnas.1222577110
75. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin (2011) 61(2):69–90. doi:10.3322/caac.20107
76. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin (2012) 62(1):10–29. doi:10.3322/caac.21149
77. Gustafsson L, Ponten J, Zack M, Adami HO. International incidence rates of invasive cervical cancer after introduction of cytological screening. Cancer Causes Control (1997) 8(5):755–63. doi:10.1023/A:1018435522475
78. Harper DM, Franco EL, Wheeler C, Ferris DG, Jenkins D, Schuind A, et al. Efficacy of a bivalent L1 virus-like particle vaccine in prevention of infection with human papillomavirus types 16 and 18 in young women: a randomised controlled trial. Lancet (2004) 364(9447):1757–65. doi:10.1016/S0140-6736(04)17398-4
79. Harper DM, Franco EL, Wheeler CM, Moscicki AB, Romanowski B, Roteli-Martins CM, et al. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet (2006) 367(9518):1247–55. doi:10.1016/S0140-6736(06)68439-0
80. Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB, et al. A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med (2002) 347(21):1645–51. doi:10.1056/NEJMoa020586
81. Mao C, Koutsky LA, Ault KA, Wheeler CM, Brown DR, Wiley DJ, et al. Efficacy of human papillomavirus-16 vaccine to prevent cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol (2006) 107(1):18–27. doi:10.1097/01.AOG.0000192397.41191.fb
82. Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, et al. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol (2005) 6(5):271–8. doi:10.1016/S1470-2045(05)70101-7
83. Greer BE, Koh WJ, Abu-Rustum NR, Apte SM, Campos SM, Chan J, et al. Cervical cancer. J Natl Compr Canc Netw (2010) 8(12):1388–416.
84. Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res (2013) 73(7):2271–80. doi:10.1158/0008-5472.CAN-12-3000
85. Tentori L, Graziani G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol Res (2005) 52(1):25–33. doi:10.1016/j.phrs.2005.02.010
86. Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res (2000) 6(7):2860–7.
87. Tentori L, Leonetti C, Scarsella M, D’Amati G, Vergati M, Portarena I, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res (2003) 9(14):5370–9.
88. Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst (2004) 96(1):56–67. doi:10.1093/jnci/djh005
89. Curtin NJ, Wang LZ, Yiakouvaki A, Kyle S, Arris CA, Canan-Koch S, et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin Cancer Res (2004) 10(3):881–9. doi:10.1158/1078-0432.CCR-1144-3
90. Cheng CL, Johnson SP, Keir ST, Quinn JA, Ali-Osman F, Szabo C, et al. Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol Cancer Ther (2005) 4(9):1364–8. doi:10.1158/1535-7163.MCT-05-0128
91. Burkle A, Chen G, Kupper JH, Grube K, Zeller WJ. Increased poly(ADP-ribosyl)ation in intact cells by cisplatin treatment. Carcinogenesis (1993) 14(4):559–61. doi:10.1093/carcin/14.4.559
92. Smith LM, Willmore E, Austin CA, Curtin NJ. The novel poly(ADP-Ribose) polymerase inhibitor, AG14361, sensitizes cells to topoisomerase I poisons by increasing the persistence of DNA strand breaks. Clin Cancer Res (2005) 11(23):8449–57. doi:10.1158/1078-0432.CCR-05-1224
93. Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, et al. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther (2003) 2(4):371–82.
94. Oza AM, Cibula D, Oaknin A, Poole CJ, Mathijssen RHJ, Sonke GS, et al. Olaparib plus paclitaxel plus carboplatin (P/C) followed by olaparib maintenance treatment in patients (pts) with platinum-sensitive recurrent serous ovarian cancer (PSR SOC): a randomized, open-label phase II study (ASCO Meeting Abstracts). J Clin Oncol (2012) 30(15):5001. [Accessed 2013 Jun 18]. Available at: http://meeting.ascopubs.org/cgi/content/abstract/30/15_suppl/
5001?sid=35889604-a227-482d-96ff-5d349c7e1856
95. Lee J, Annunziata CM, Minasian LM, Zujewski J, Prindiville SA, Kotz HL, et al. Phase I study of the PARP inhibitor olaparib (O) in combination with carboplatin (C) in BRCA1/2 mutation carriers with breast (Br) or ovarian (Ov) cancer (Ca) (ASCO Meeting Abstracts). J Clin Oncol (2011) 29(15):2520. [Accessed 2013 Jun 10]. Available at: http://meeting.ascopubs.org/cgi/content/short/29/15_suppl/2520?rss=1
96. Yu W, Gu K, Yu Z, Yuan D, He M, Ma N, et al. Sorafenib potentiates irradiation effect in hepatocellular carcinoma in vitro and in vivo. Cancer Lett (2013) 329(1):109–17. doi:10.1016/j.canlet.2012.10.024
97. Yadav A, Kumar B, Teknos TN, Kumar P. Sorafenib enhances the antitumor effects of chemoradiation treatment by downregulating ERCC-1 and XRCC-1 DNA repair proteins. Mol Cancer Ther (2011) 10(7):1241–51. doi:10.1158/1535-7163.MCT-11-0004
98. Rajesh M, Mukhopadhyay P, Batkai S, Godlewski G, Hasko G, Liaudet L, et al. Pharmacological inhibition of poly(ADP-ribose) polymerase inhibits angiogenesis. Biochem Biophys Res Commun (2006) 350(2):352–7. doi:10.1016/j.bbrc.2006.09.160
99. Pyriochou A, Olah G, Deitch EA, Szabo C, Papapetropoulos A. Inhibition of angiogenesis by the poly(ADP-ribose) polymerase inhibitor PJ-34. Int J Mol Med (2008) 22(1):113–8.
100. Tentori L, Lacal PM, Muzi A, Dorio AS, Leonetti C, Scarsella M, et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur J Cancer (2007) 43(14):2124–33. doi:10.1016/j.ejca.2007.07.010
101. Liu J, Fleming GF, Tolaney SM, Birrer MJ, Penson RT, Berlin ST, et al. A phase I trial of the PARP inhibitor olaparib (AZD2281) in combination with the antiangiogenic cediranib (AZD2171) in recurrent ovarian or triple-negative breast cancer (ASCO Meeting Abstracts). J Clin Oncol (2011) 29(15):5028. [Accessed 2013 Jun 10]. Available at: http://meeting.ascopubs.org/cgi/content/abstract/29/15_suppl/
5028?sid=a88c5b0a-3c23-4d7b-893a-877d2132a7db
102. Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res (2003) 63(18):6008–15.
103. de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A (1997) 94(14):7303–7. doi:10.1073/pnas.94.14.7303
104. Masutani M, Nozaki T, Nishiyama E, Shimokawa T, Tachi Y, Suzuki H, et al. Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice. Mol Cell Biochem (1999) 193(1-2):149–52. doi:10.1023/A:1006941016799
105. Veliparib with radiation therapy in patients with inflammatory or loco-regionally recurrent breast cancer. (2013). [Accessed 2013 June 18]. Available from: http://clinicaltrials.gov/ct2/show/NCT01477489?term=parp+inhibitors+
and+radiation+and+breast+cancer&rank=1
106. Study of the poly (ADP-ribose) polymerase-1 (PARP-1) inhibitor BSI-201 in patients with newly diagnosed malignant glioma. (2013). [Accessed 2013 Jun 18]. Available from: http://clinicaltrials.gov/ct2/show/NCT00687765?term=glioblastoma+multiforme
+and+parp+inhibitors+and+radiation&rank=1
107. Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res (2010) 16(8):2344–51. doi:10.1158/1078-0432.CCR-09-2758
108. Mukhopadhyay A, Plummer ER, Elattar A, Soohoo S, Uzir B, Quinn JE, et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res (2012) 72(22):5675–82. doi:10.1158/0008-5472.CAN-12-0324
109. Ji J, Kinders RJ, Zhang Y, Rubinstein L, Kummar S, Parchment RE, et al. Modeling pharmacodynamic response to the poly(ADP-ribose) polymerase inhibitor ABT-888 in human peripheral blood mononuclear cells. PLoS ONE (2011) 6(10):e26152. doi:10.1371/journal.pone.0026152
110. Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res (2012) 18(6):1726–34. doi:10.1158/1078-0432.CCR-11-2821
111. Do K, Chen AP. Molecular pathways: targeting PARP in cancer treatment. Clin Cancer Res (2013) 19(5):977–84. doi:10.1158/1078-0432.CCR-12-0163
112. Ang J, Yap TA, Fong P, Carden CP, Tan DS, Hanwell J, et al. Preliminary experience with the use of chemotherapy (CT) following treatment with olaparib, a poly(ADP-ribose) polymerase inhibitor (PARPi), in patients with BRCA1/2-deficient ovarian cancer (BDOC) (ASCO Meeting Abstracts). J Clin Oncol (2010) 28(15):5041. [Accessed 6/10/2013]. Available from: http://meeting.ascopubs.org/cgi/content/abstract/28/15_suppl/5041
113. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A (2008) 105(44):17079–84. doi:10.1073/pnas.0806092105
Keywords: ovarian cancer, cervical cancer, endometrial cancer, PARP inhibitor, olaparib, veliparib, rucaparib, niraparib
Citation: Reinbolt RE and Hays JL (2013) The role of PARP inhibitors in the treatment of gynecologic malignancies. Front. Oncol. 3:237. doi: 10.3389/fonc.2013.00237
Received: 25 June 2013; Paper pending published: 16 July 2013;
Accepted: 28 August 2013; Published online: 01 October 2013.
Edited by:
Kristin Zorn, Magee-Womens Hospital of UPMC, USAReviewed by:
Maria Felice Brizzi, University of Torino, ItalyJian Lu, Johns Hopkins University, USA
Copyright: © 2013 Reinbolt and Hays. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John L. Hays, Division of Medical Oncology, The James Comprehensive Cancer Center, 588 Biomedical Research Tower, 460 W 12th Avenue, Columbus, OH 43210, USA e-mail: john.hays@osumc.edu