The Impact of Maternal High-Fat Diet on Bone Microarchitecture in Offspring
 Emma J. Buckels
Emma J. Buckels Scott M. Bolam
Scott M. Bolam Mei Lin Tay
Mei Lin Tay Brya G. Matthews
Brya G. Matthews- 1Department of Molecular Medicine and Pathology, School of Medical Sciences, University of Auckland, Auckland, New Zealand
- 2Department of Surgery, School of Medicine, University of Auckland, Auckland, New Zealand
- 3Department of Orthopaedic Surgery, Auckland City Hospital, Auckland, New Zealand
The incidence of obesity in women of reproductive age has significantly increased over the past 100 years. There is a well-established connection between maternal obesity during pregnancy and an increased risk of developing non-communicable cardiometabolic diseases in her offspring. This mini-review focuses on evidence examining the effect of maternal high-fat diet (HFD) on skeletal development and bone health in later life in offspring. The majority of rodent studies indicate that maternal HFD generally negatively affects both embryonic bone development and bone volume in adult animals. Details surrounding the mechanisms of action that drive changes in the skeleton in offspring remain unclear, although numerous studies suggest that some effects are sex-specific. Human studies in this area are limited but also suggest that HFD during pregnancy may impair bone formation and increase fracture risk during childhood. Given the consequences of low bone mass and deranged bone microarchitecture for offspring, advances in our understanding of the developmental origins of bone health is critical in the battle against osteoporosis.
Introduction
The prevalence of obesity over the past 100 years has dramatically increased, with obesity identified as the most common metabolic disorder. Globally, an estimated 600 million adults were obese (body mass index ≥ 30 kg/m2), and 1.9 billion adults were overweight in 2015 (body mass index 25–30 kg/m2) (1). The prevalence of obesity is expected to reach 1.12 billion individuals by 2030 (2). Obese individuals have an increased risk of morbidity from type 2 diabetes mellitus (T2DM), cardiovascular disease, specific cancers, and osteoarthritis (3).
The incidence of obesity in women of reproductive age has also increased. Maternal obesity is a significant risk factor for maternal, fetal, and neonatal morbidities, including miscarriage, preterm delivery, hypertension, pre-eclampsia, and gestational diabetes (4–6). Research in the field of developmental origins of health and disease (DOHaD) has highlighted that maternal obesity during pregnancy predisposes offspring to develop obesity and other non-communicable diseases, including T2DM, hypertension, and cardiovascular disease, later in adulthood (7). Paternal obesity also increases the risk of developing non-communicable diseases in offspring (8–10), and both maternal and paternal obesity have transgenerational effects on subsequent generations via epigenetic effects on the germline (10–12). Thus, obesity and its related comorbidities represent an increasing burden on healthcare systems.
Although not fully understood, the effect of maternal obesity on the development of various organs and tissues such as the brain, liver, kidney, endocrine pancreas, and skeletal muscle and their structure and function have been well-researched with the aid of animal models. In these models, maternal obesity is induced via a high-fat diet (HFD) (13, 14). Recently the effects of maternal HFD on bone mass and strength in offspring and the risk of developing osteoporosis later in life have been researched. This mini-review will discuss evidence that maternal HFD-induced obesity affects bone development and microarchitecture, focusing on recent advancements using rodent models, and will discuss the potential mechanisms involved.
Early Bone Development and Impact Later in Life
The skeleton develops in utero from mesenchymal condensations. Most of the skeleton forms via a process known as endochondral ossification: initially, a cartilaginous template forms, which is later progressively replaced by mineralized bone matrix. Cartilaginous growth plates continue to control the longitudinal growth of bones throughout neonatal and childhood growth, while the overall bone shape, mineralization, and microarchitecture are determined by the balance of bone formation by osteoblasts and bone resorption by osteoclasts in different locations. Longitudinal skeletal growth continues until the late teens in humans, ending when the growth plates fuse. However, bone mineral density (BMD) continues to increase slowly until peak bone mass is reached in the mid-20s to early-30s. Rodents continue to grow slowly for a longer portion of their life, but mice reach peak bone mass at ~12-weeks of age. Bone mass begins to decline as bone resorption outpaces bone formation as we age. In women, there is a dramatic period of bone loss following menopause when reductions in sex hormone levels affect homeostasis. Skeletal size, BMD, and bone microarchitecture are largely determined by genetics, with up to 85% of the variation in BMD explained by genetic factors (15). However, various non-genetic factors also influence both bone accrual during growth and bone loss in later life. Nutrition is a major factor influencing both growth and bone mass and can have effects at all life stages. Exercise, or the effect of loading on the skeleton, plays a major role in bone accrual and retention.
Osteoporosis is characterized by low BMD and a high risk of fracture, and affects one-half of elderly women and about one-fifth of elderly men. While osteoporosis is considered a disease of aging, early life events and a failure to achieve maximal peak bone mass determined by an individual's genetics can significantly impact future osteoporosis risk. One line of evidence for long-term impacts of events earlier in life comes from studies in athletes. Baseball pitchers who develop larger, stronger bones in their throwing arm during early adulthood can retain better bone structure in this arm for 50 years following retirement from the game (16). Various drugs are available that effectively reduce fracture risk in people with osteoporosis; however, the majority are antiresorptive therapies that prevent further bone loss but do not enable the replacement of bone already lost (17). Anabolic treatments restore bone microarchitecture to some degree, but are expensive biologics so not available to everyone. Understanding risk factors for low bone mass and identifying people at high risk of fracture are important for preventing fracture-related morbidity and mortality in aging populations.
Effect of Maternal HFD on Bone in Rodents
Thirteen rodent studies considered the effects of maternal HFD on offspring bone development (Table 1). The majority of studies implemented the maternal HFD regime before mating (4–15 weeks before conception) and continued through pregnancy and lactation. However, in three studies, the maternal HFD-feeding window was exclusively during pregnancy and lactation and exclusively during lactation in one study.
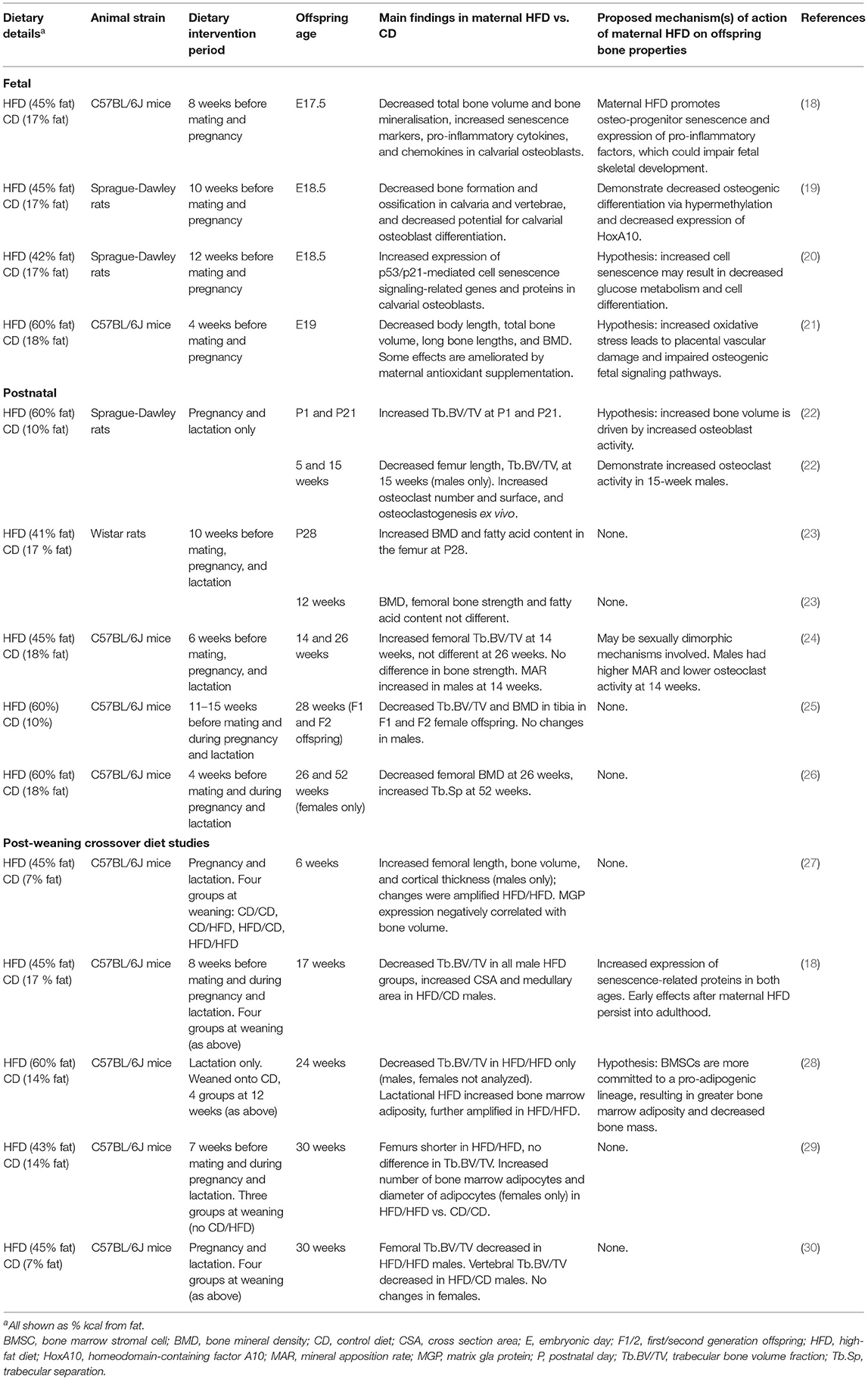
Table 1. Studies investigating the effect of maternal HFD on offspring bone properties.
Fetal and Neonatal Offspring
Offspring of dams fed a maternal HFD before and during pregnancy have evidence of skeletal developmental delay in late-gestation with decreased bone formation, bone volume, and BMD (18–21). Chen et al. have demonstrated that maternal HFD promotes cellular senescence in fetal calvarial osteoblasts cells, potentially suppressing fetal bone formation (18–20).
In both newborns and weanlings, exposure to maternal HFD resulted in increased total bone mass, BMD, and trabecular bone volume in long bones (22, 23). This phenotype is likely the result of increased osteoblast activity, as bone modeling is most active over this period of rapid growth (31). Increased bone mass in weanlings may be an indirect effect of maternal HFD, as these offspring consume more milk, and the milk consumed has a higher energy content compared to dams fed control diet (CD) (22). Additionally, Miotto et al. found higher concentrations of monounsaturated fatty acids in the long bones of offspring exposed to maternal HFD, which is likely to reflect the diet consumed by the dams; these lipid stores may have supported rapid bone growth (23).
Adult Offspring
From early adulthood, a pattern of sustained bone loss in offspring of dams fed HFD is reported. In most studies, offspring exposed to maternal HFD have reduced BMD and bone volume in long bones and vertebrae from as early as 8-weeks of age, which persisted over their lifetime (18, 21–23, 25, 30). Hafner et al. found that maternal HFD during lactation alone was sufficient to increase bone marrow adiposity (28). Maternal HFD decreased trabecular bone parameters in offspring (18, 21–23, 25, 30). However, this effect was not observed in all studies; two studies found increased femoral bone trabecular volume and increased cortical thickness following maternal HFD (24, 27). Notably, these two studies analyzed offspring who were young adults, as opposed to the studies that found decreased bone volume in mice who were considerably older.
Several studies demonstrated sex-specific variation in the effect of maternal HFD, with males more likely to exhibit a bone phenotype than females (18, 22, 24). Only one study exclusively found changes in bones in females following maternal HFD (25). Therefore, maternal HFD most likely has a sexually dimorphic effect on the skeleton of offspring.
Multigenerational Effects of Maternal HFD
Maternal HFD can have multigenerational effects on bone in offspring and grand-offspring. Harasymowicz et al. found decreased trabecular bone volume and BMD in F1 and F2 generations, even with no additional exposure to HFD in either generation (25).
Postnatal Exposure to HFD
A post-weaning HFD, or a “second hit,” has the potential to amplify the effects of maternal HFD (32). Three studies that investigated continued feeding of HFD in offspring after weaning found that post-weaning HFD further decreased trabecular bone volume (28, 30) or increased bone-marrow adiposity (28, 29), compared to exposure to maternal HFD alone.
Effect of Maternal Obesity and HFD on Bone in Humans
Several studies have specifically addressed whether maternal obesity or maternal HFD during pregnancy affects bone development in offspring, both in utero and post-partum (33). Longitudinal studies show that obese mothers have babies with increased body length, whole-body bone area, and mineral content (34–36), but maternal diet was not reported. Two studies demonstrated that mothers consuming a high-fat “Western diet” during pregnancy, defined as a diet high in meat, processed food, and saturated fat, have children with lower whole-body bone area, bone mineral content, and BMD, compared with children of mothers on low-fat “prudent diets” during pregnancy, defined as a diet high in fruits, vegetables, grains and low-fat dairy products (37, 38). Interestingly, offspring of mothers in the Danish National Birth Cohort who consumed a Western diet had a significantly increased risk of fracture between birth and 16-years of age (39). None of these studies reported maternal BMI related to study groups. Due to the paucity of human data (33), it is unclear whether maternal obesity in the absence of HFD, maternal HFD in the absence of obesity, or any other dietary conditions of over-nutrition with or without maternal obesity affects the skeletal phenotype in human offspring.
Mechanisms of Action
Research into the mechanisms involved in linking maternal HFD with a bone phenotype in rodents remains in its infancy, and to our knowledge, no studies in humans have explored any mechanisms of action. The following section discusses some key mechanisms demonstrated in rodent maternal HFD studies, linking the early life environment and the observed bone phenotype in these offspring (Figure 1).
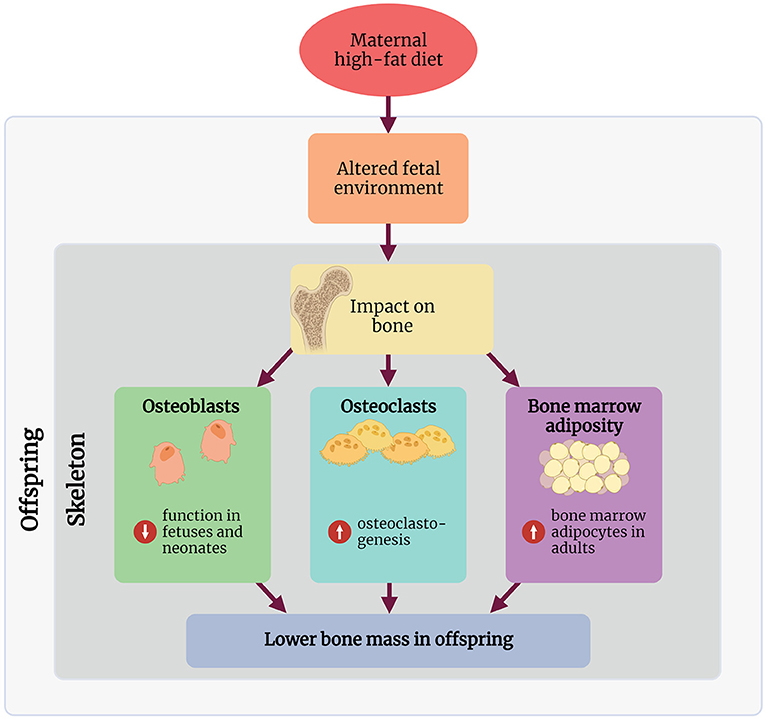
Figure 1. Summary of the proposed mechanisms through which maternal high-fat diet affects the skeleton in the offspring. Created with BioRender.com.
Osteoblasts
Osteoblasts are derived from mesenchymal stem cells and are responsible for the synthesis and mineralisation of bone. Whilst osteoblast number is unaffected (22, 24), there may be a negative relationship between osteoblast function in offspring and maternal HFD during pregnancy and lactation. However, this relationship with osteoblast function may be transient and lost as offspring age. Whole-embryo skeletal ossification and total bone volume are decreased following maternal HFD (18, 19). Rat calvarial osteoblasts from offspring exposed to maternal HFD have decreased proliferation and osteoblastic differentiation (19). Therefore, decreased differentiation of osteoblasts could directly be responsible for decreased or delayed bone formation during development.
Although bone marrow stromal cells (BMSCs) are a significant source of osteoblast progenitor cells contributing to bone remodeling, only one study examined whether the differentiation capacity of these cells into osteoblasts is influenced by maternal HFD. Kushwaha et al. assessed cellular activity in 15-week old animals and found no difference in osteogenic differentiation of BMSCs exposed to maternal HFD. However, compared to BMSCs derived from CD-fed mothers, these cells have higher mRNA expression of RANKL, which will have implications for osteoclastogenesis (22).
Osteoblast function in adult rodents exposed to maternal HFD is variable. Circulating osteocalcin concentrations are decreased at 17-weeks of age in mice, indicating osteoblast function is decreased following maternal HFD. Mineral apposition rate (MAR) is a reliable direct measurement of osteoblast function (40); one rat study found no difference in MAR in 15-week old males following maternal HFD (22). Interestingly, Devlin et al. found that MAR is increased in 14-week old male mice exposed to maternal HFD (24). This relationship is no longer detected in male mice at 26-weeks of age (24), indicating the rate of mineralization has decreased to a level similar to maternal CD offspring. Surprisingly, Devlin et al. also found no difference in concentrations of the bone formation marker type 1 procollagen N-terminal (P1NP) in circulation at either 14- or 26-weeks of age, despite observed differences in MAR (24). Overall, there is no consensus on whether maternal HFD affects osteoblast function in adult offspring.
Osteoclasts
Osteoclasts are multinucleated phagocytic cells responsible for bone resorption and are derived from the macrophage-monocyte cell lineage. Very few studies have considered the effects of maternal HFD on osteoclasts. One study broadly examined ex vivo osteoclastogenesis following maternal HFD during pregnancy and lactation (22). Kushwaha et al. demonstrated via histomorphometry that osteoclast number, erosion surface, and osteoclast surface were increased in 15-week old male rats exposed to maternal HFD. Ex vivo cultures of osteoclast precursors isolated from these animals had increased potential to differentiate into osteoclasts, with these osteoclasts more numerous and larger. Interestingly, these osteoclasts were more sensitized to the effects of RANKL and had increased RANK mRNA expression. Cultured osteoblasts from these same animals had increased RANKL mRNA expression, indicating that the potential for osteoclastogenesis is increased following maternal HFD. Alternatively, when mice were fed HFD for 6-weeks before mating, pregnancy, and lactation, their offspring demonstrated no significant difference in osteoclast number but decreased osteoclast activity at 14- and 26-weeks of age (24). Increased bone resorption is a major contributor to decreased bone volume that develops when rodents are fed HFD, which is likely secondary to increased inflammation (41). Maternal HFD is known to cause low-grade chronic inflammation in offspring; therefore, it is feasible that this could contribute to bone loss in these animals (42). Chen et al. noted increased inflammatory cytokine production in fetal calvarial osteoblasts exposed to maternal HFD, but this potential mechanism has not been addressed in adult offspring (18). Given these conflicting data, further studies are needed to determine the effects of maternal HFD on osteoclast number and function.
Bone Marrow Adiposity
The balance between BMSCs giving rise to osteogenic or adipogenic precursors is critical for maintaining bone mass; if this balance is shifted toward adipogenesis, this may come at the expense of osteoblastogenesis (43). Additionally, increased bone-marrow adiposity can affect osteogenesis, with an apparent negative relationship between bone marrow adiposity and bone mass (44). Maternal HFD during pregnancy and lactation (29) or lactation only (28) is associated with increased adipocyte number and adipocyte size in the bone marrow cavity. However, interpretation of these studies is complicated by their study design; offspring were either weaned directly onto HFD or CD, or onto CD followed by HFD between 12 and 24-weeks of age. Both studies had conflicting results as to whether maternal HFD with post-weaning HFD affected bone microarchitecture. Hafner et al. found bone marrow adiposity was increased, and trabecular bone volume was decreased in the maternal HFD/post-weaning HFD group at 24-weeks of age (28). However, Lanham et al. found no difference in bone microarchitecture at 30-weeks of age (29). No studies performed ex vivo adipogenesis assays on BMSCs. Further studies are required to confirm whether changes in bone marrow adiposity contribute to the bone phenotype in these offspring.
Epigenetic Modifications
Epigenetic modifications, including DNA methylation and various post-translational histone modifications, describe changes to gene expression that occur without affecting the underlying DNA sequence. Epigenetic modifications allow the individual to alter gene expression in response to the environment and have long been considered a principal mechanism through which the early-life environment affects offspring (7, 45). Despite this connection, there is a paucity of studies that have specifically measured epigenetic modifications in response to maternal HFD.
Chen et al. demonstrated that maternal HFD promotes cellular senescence in fetal calvarial osteoblasts, potentially suppressing bone formation in the prenatal period in both mice and rats (18–20). These findings were shown to be through increased expression of p300/CBP, which increased H3K27 acetylation, which promoted p53/p21-mediated cell senescence signaling in pre-osteoblasts; increased expression of p300/CPB persisted until adulthood (18). Maternal HFD also promoted increases in methylated CpG sites in the homeobox protein A10 (HoxA10) promoter. HoxA10 is important for fetal osteoblastogenesis and adult bone regeneration.
Current Research Gaps
Many unanswered questions surrounding how HFD-induced maternal obesity affects bone development and microarchitecture in offspring remain.
One outstanding question is whether the detrimental effects on offspring skeleton are driven by maternal obesity, maternal HFD, or both. In humans, most studies explore the effects of obesity during pregnancy, commonly assessed by measuring body mass index rather than dietary patterns (46). In rodents, the majority of studies implemented a maternal HFD regime at least 4 weeks before mating. This would have induced an obesity phenotype in these dams, as well as ongoing exposure to HFD. However, it is unlikely that maternal obesity would have been induced in studies where HFD-feeding was restricted to pregnancy and lactation, or lactation alone. Therefore, these offspring likely experienced exposure to HFD in the absence of maternal obesity. It is challenging to tease out whether HFD-induced obesity before pregnancy or ongoing maternal HFD affects the skeleton in offspring using a rodent model. Unlike in humans, changing the diet of a rodent from HFD to CD induces rapid weight loss (47). This could be overcome using embryo transfer following pre-conception maternal HFD, placing embryos into CD-fed recipients (46).
Another gap in our understanding is deciphering the effects of maternal HFD on other tissues in offspring and how these effects, in turn, modulate the skeleton. For instance, there is cross-talk between the skeleton and skeletal muscle, adipose tissue, and the endocrine pancreas (48–52). The structure and function of these tissues are affected by the early life environment (7). Therefore, it would be interesting to determine whether this cross-talk is affected by maternal HFD and the downstream effects on the skeleton.
In this mini-review, we exclusively discussed the effect of maternal HFD on the skeleton in offspring; however, other paradigms of early life exposure to nutritional excess are also worthy of exploration. For instance, a maternal high-protein diet (53) and a combination of high-fat and high-sugar diet (54) also negatively impact the skeleton in offspring. Additionally, pre-conception paternal nutrition also has long-term effects on the metabolic health of offspring (55). We are unaware of any studies that have addressed the paternal influence of skeletal development in offspring. Thus, understanding the impact of paternal health will also be necessary for understanding the mechanisms that link the early life environment with skeletal health in offspring.
Critically, we are unaware of any studies investigating the effect of nutritional, pharmacological, or behavioral interventions on skeletal outcomes in the offspring. Whilst we do not yet fully understand the mechanisms that impact the developing skeleton in response to maternal HFD, a significant gap lies in the lack of intervention studies.
Conclusion
There is growing evidence that exposure to maternal HFD during pregnancy has long-lasting adverse effects on the skeleton of offspring. However, many details surrounding these changes and the mechanisms of action that drive these effects remain unclear, and further basic studies are required. Given the consequences of low bone mass and deranged bone microarchitecture for offspring, advances in our understanding of the developmental origins of bone health is critical in our battle against diseases like osteoporosis.
Author Contributions
EB designed the review. EB, SB, and MT collected relevant articles. All authors have contributed to writing and revision of the manuscript, read, and approved the submitted version.
Funding
This research was funded by Health Research Council of New Zealand Sir Charles Hercus Fellowship, American Society of Bone and Mineral Research Rising Star Award, Auckland Medical Research Foundation Project Grant 1118008 to BM. The University of Auckland Doctoral Scholarship to MT. Health Research Council Clinical Research Training Fellowship to SB.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Marcus Ground for assistance with drawing the diagram.
References
1. Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metab Clin Exp. (2019) 92:6–10. doi: 10.1016/j.metabol.2018.09.005
2. Kelly T, Yang W, Chen C-S, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obesity. (2008) 32:1431–7. doi: 10.1038/ijo.2008.102
3. Apovia CM. Obesity: definition, comorbidities, causes, and burden. Am J Manag Care. (2016) 22.7:s176–85.
4. Davies GA, Maxwell C, McLeod L, Gagnon R, Basso M, Bos H, et al. Obesity in pregnancy. J Obstetr Gynaecol Canada. (2010) 32:165–73. doi: 10.1016/S1701-2163(16)34432-2
5. Sirimi N, Goulis DG. Obesity in pregnancy. Hormones. (2010) 9:299–306. doi: 10.14310/horm.2002.1280
6. Catalano PM. Obesity, insulin resistance and pregnancy outcome. Reproduction. (2010) 140:365–71. doi: 10.1530/REP-10-0088
7. Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood disease. Biochem J. (2010) 427:333–47. doi: 10.1042/BJ20091861
8. Fernandez-Twinn DS, Hjort L, Novakovic B, Ozanne SE, Saffery R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia. (2019) 62:1789–801. doi: 10.1007/s00125-019-4951-9
9. Eriksson JG. Developmental pathways and programming of diabetes: epidemiological aspects. J Endocrinol. (2019) 242:T95–104. doi: 10.1530/JOE-18-0680
10. Hur SS, Cropley JE, Suter CM. Paternal epigenetic programming: evolving metabolic disease risk. J Mol Endocrinol. (2017) 58:R159–68. doi: 10.1530/JME-16-0236
11. Vickers MH. Developmental programming and transgenerational transmission of obesity. Ann Nutr Metab. (2014) 64:26–34. doi: 10.1159/000360506
12. Drake AJ, Liu L. Intergenerational transmission of programmed effects: public health consequences. Trends Endocrinol Metab. (2010) 21:206–13. doi: 10.1016/j.tem.2009.11.006
13. Desai M, Jellyman JK, Han G, Beall M, Lane RH, Ross MG. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am J Obstetr Gynecol. (2014) 211:e1–13. doi: 10.1016/j.ajog.2014.03.025
14. Williams L, Seki Y, Vuguin PM, Charron MJ. Animal models of in utero exposure to a high fat diet: a review. Biochim Biophys Acta. (2014) 1842:507–19. doi: 10.1016/j.bbadis.2013.07.006
15. Boudin E, Fijalkowski I, Hendrickx G, Van Hul W. Genetic control of bone mass. Mol Cell Endocrinol. (2016) 432:3–13. doi: 10.1016/j.mce.2015.12.021
16. Warden SJ, Roosa SMM, Kersh ME, Hurd AL, Fleisig GS, Pandy MG, et al. Physical activity when young provides lifelong benefits to cortical bone size and strength in men. Proc Natl Acad Sci USA. (2014) 111:5337–42. doi: 10.1073/pnas.1321605111
17. Seeman E, Martin TJ. Antiresorptive and anabolic agents in the prevention and reversal of bone fragility. Nat Rev Rheumatol. (2019) 15:225–36. doi: 10.1038/s41584-019-0172-3
18. Chen J-R, Lazarenko OP, Zhao H, Alund AW, Shankar K. Maternal obesity impairs skeletal development in adult offspring. J Endocrinol. (2018) 239:33–47. doi: 10.1530/JOE-18-0244
19. Chen J-R, Zhang J, Lazarenko OP, Kang P, Blackburn ML, Ronis MJJ, et al. Inhibition of fetal bone development through epigenetic down-regulation of HoxA10 in obese rats fed high-fat diet. FASEB J. (2012) 26:1131–41. doi: 10.1096/fj.11-197822
20. Chen J-R, Lazarenko OP, Blackburn ML, Rose S, Frye RE, Badger TM, et al. Maternal obesity programs senescence signaling and glucose metabolism in osteo-progenitors from rat and human. Endocrinology. (2016) 157:4172–83. doi: 10.1210/en.2016-1408
21. Liang C, Oest ME, Jones JC, Prater MR. Gestational high saturated fat diet alters C57BL/6 mouse perinatal skeletal formation. Birth Defects Res. (2009) 86:362–9. doi: 10.1002/bdrb.20204
22. Kushwaha P, Khambadkone SG, Li M, Goodman EJ, Aravindan N, Riddle RC, et al. Maternal high-fat diet induces long-lasting defects in bone structure in rat offspring through enhanced osteoclastogenesis. Calcified Tissue Int. (2021) 108:680–92. doi: 10.1007/s00223-020-00801-4
23. Miotto PM, M CL, Amoye F, LeBlanc PJ, Peters SJ, Roy BD, et al. Maternal high fat feeding does not have long-lasting effects on body composition and bone health in female and male Wistar rat offspring at young adulthood. Molecules. (2013) 18:15094–109. doi: 10.3390/molecules181215094
24. Devlin MJ, Grasemann C, Cloutier AM, Louis L, Alm C, Palmert MR, et al. Maternal perinatal diet induces developmental programming of bone architecture. J Endocrinol. (2013) 217:69–81. doi: 10.1530/JOE-12-0403
25. Harasymowicz NS, Choi Y-R, Wu C-L, Iannucci L, Tang R, Guilak F. Intergenerational transmission of diet-induced obesity, metabolic imbalance, and osteoarthritis in mice. Arthrit Rheumatol. (2020) 72:632–44. doi: 10.1002/art.41147
26. Liang C, Oest ME, Prater MR. Intrauterine exposure to high saturated fat diet elevates risk of adult-onset chronic diseases in C57BL/6 mice. Birth Defects Res. (2009) 86:377–84. doi: 10.1002/bdrb.20206
27. Lanham SA, Cagampang FR, Oreffo ROC. Maternal high-fat diet and offspring expression levels of vitamin K-dependent proteins. Endocrinology. (2014) 155:4749–61. doi: 10.1210/en.2014-1188
28. Hafner H, Chang E, Carlson Z, Zhu A, Varghese M, Clemente J, et al. Lactational high-fat diet exposure programs metabolic inflammation and bone marrow adiposity in male offspring. Nutrients. (2019) 11:1393. doi: 10.3390/nu11061393
29. Lanham SA, Roberts C, Hollingworth T, Sreekumar R, Elahi MM, Cagampang FR, et al. Maternal high-fat diet: effects on offspring bone structure. Osteopor Int. (2010) 21:1703–14. doi: 10.1007/s00198-009-1118-4
30. Lanham SA, Cagampang FR, Oreffo ROC. Maternal high fat diet affects offspring's vitamin K-dependent proteins expression levels. PLoS ONE. (2015) 10:e0138730. doi: 10.1371/journal.pone.0138730
31. Kimmel DB, Jee WSS. Bone cell kinetics during longitudinal bone growth in the rat. Calcified Tissue Int. (1980) 32:123–33. doi: 10.1007/BF02408531
32. Dickinson H, Moss TJ, Gatford KL, Moritz KM, Akison L, Fullston T, et al. A review of fundamental principles for animal models of DOHaD research: an Australian perspective. J Dev Origins Health Dis. (2016) 7:449–72. doi: 10.1017/S2040174416000477
33. Jensen KH, Riis KR, Abrahamsen B, Handel MN. Nutrients, diet, and other factors in prenatal life and bone health in young adults: a systematic review of longitudinal studies. Nutrients. (2020) 12:2866. doi: 10.3390/nu12092866
34. Enstad S, Cheema S, Thomas R, Fichorova RN, Martin CR, O'Tierney-Ginn P, et al. The impact of maternal obesity and breast milk inflammation on developmental programming of infant growth. Eur J Clin Endocrinol. (2021) 75:180–8. doi: 10.1038/s41430-020-00720-5
35. Harvey NC, Javaid MK, Arden NK, Poole JR, Crozier SR, Robinson SM, et al. Maternal predictors of neonatal bone size and geometry: the Southampton Women's Survey. J Dev Origins Health Dis. (2010) 1:35–41. doi: 10.1017/S2040174409990055
36. Zhang C, Hediger ML, Albert PS, Grewal J, Sciscione A, Grobman WA, et al. Association of maternal obesity with longitudinal ultrasonographic measures of fetal growth: findings from the NICHD fetal growth studies–singletons. JAMA Pediatr. (2018) 172:24–31. doi: 10.1001/jamapediatrics.2017.3785
37. Cole ZA, Gale CR, Javaid MK, Robinson SM, Law C, Boucher BJ, et al. Maternal dietary patterns during pregnancy and childhood bone mass: a longitudinal study. J Bone Mineral Res. (2009) 24:663–8. doi: 10.1359/jbmr.081212
38. Yin J, Dwyer T, Cochrane J, Jones G. The association between maternal diet during pregnancy and bone mass of the children at age 16. Eur J Clin Nutr. (2010) 64:131–7. doi: 10.1038/ejcn.2009.117
39. Petersen SB, Rasmussen MA, Olsen SF, Vestergaard P, Mølgaard C, Halldorsson TI, et al. Maternal dietary patterns during pregnancy in relation to offspring forearm fractures: prospective study from the Danish National Birth Cohort. Nutrients. (2015) 7:2382–400. doi: 10.3390/nu7042382
40. Recker RR, Kimmel DB, Dempster D, Weinstein RS, Wronski TJ, Burr DB. Issues in modern bone histomorphometry. Bone. (2011) 49:955–64. doi: 10.1016/j.bone.2011.07.017
41. Shu L, Beier E, Sheu T, Zhang H, Zuscik MJ, Puzas EJ, et al. High-fat diet causes bone loss in young mice by promoting osteoclastogenesis through alteration of the bone marrow environment. Calcified Tissue Int. (2015) 96:313–23. doi: 10.1007/s00223-015-9954-z
42. Zhou D, Pan YX. Pathophysiological basis for compromised health beyond generations: role of maternal high-fat diet and low-grade chronic inflammation. J Nutr Biochem. (2015) 26:1–8. doi: 10.1016/j.jnutbio.2014.06.011
43. Rharass T, Lucas S. Mechanisms in endocrinology: bone marrow adiposity and bone, a bad romance? Eur J Endocrinol. (2018) 179:R165–82. doi: 10.1530/EJE-18-0182
44. Devlin MJ, Rosen CJ. The bone-fat interface: basic and clinical implications of marrow adiposity. Lancet Diabetes Endocrinol. (2015) 3:141–7. doi: 10.1016/S2213-8587(14)70007-5
45. Bansal A, Simmons RA. Epigenetics and developmental origins of metabolic dysfunction: correlation or causation? Am J Physiol Endocrinol Metab. (2018) 315:E15–28. doi: 10.1152/ajpendo.00424.2017
46. Christians JK, Lennie KI, Wild LK, Garcha R. Effects of high-fat diets on fetal growth in rodents: a systematic review. Reprod Biol Endocrinol. (2019) 17:39. doi: 10.1186/s12958-019-0482-y
47. Matikainen-Ankney BA, Ali MA, Miyazaki NL, Fry SA, Licholai JA, Kravitz AV. Weight loss after obesity is associated with increased food motivation and faster weight regain in mice. Obesity. (2020) 28:851–6. doi: 10.1002/oby.22758
48. Ducy P. The role of osteocalcin in the endocrine cross-talk between bone remodelling and energy metabolism. Diabetologia. (2011) 54:1291–7. doi: 10.1007/s00125-011-2155-z
49. Sims NA, Walsh NC. Intercellular cross-talk among bone cells: new factors and pathways. Curr Osteopor Rep. (2012) 10:109–17. doi: 10.1007/s11914-012-0096-1
50. Argiles JM, Lopez-Soriano J, Almendro V, Busquets S, López-Soriano FJ. Cross-talk between skeletal muscle and adipose tissue: a link with obesity? Med Res Rev. (2004) 25:49–65. doi: 10.1002/med.20010
51. Brotto M, Johnson ML. Endocrine crosstalk between muscle and bone. Curr Osteopor Rep. (2014) 12:135–41. doi: 10.1007/s11914-014-0209-0
52. Li F, Li Y, Duan Y, Hu C-AA, Tang Y, Yin Y. Myokines and adipokines: involvement in the crosstalk between skeletal muscle and adipose tissue. Cytok Growth Fact Rev. (2017) 33:73–82. doi: 10.1016/j.cytogfr.2016.10.003
53. Ellur G, Sukhdeo SV, Khan MT, Sharan K. Maternal high protein-diet programs impairment of offspring's bone mass through miR-24-1-5p mediated targeting of SMAD5 in osteoblasts. Cell Mol Life Sci. (2021) 78:1729–44. doi: 10.1007/s00018-020-03608-6
54. Shi Y, Saben JL, He G, Moley KH, Long F. Diet-induced metabolic dysregulation in female mice causes osteopenia in adult offspring. J Endocr Soc. (2020) 4:1–14. doi: 10.1210/jendso/bvaa028
Keywords: early life nutrition, maternal obesity, developmental origins of health and disease (DOHaD), osteoporosis, osteoblast, osteoclast, bone marrow adipocytes, skeletal development
Citation: Buckels EJ, Bolam SM, Tay ML and Matthews BG (2021) The Impact of Maternal High-Fat Diet on Bone Microarchitecture in Offspring. Front. Nutr. 8:730037. doi: 10.3389/fnut.2021.730037
Received: 24 June 2021; Accepted: 02 August 2021;
Published: 30 August 2021.
Edited by:
Clare Marie Reynolds, University College Dublin, IrelandReviewed by:
Tania Romano, La Trobe University, AustraliaDian Teguh, Beth Israel Deaconess Medical Center and Harvard Medical School, United States
Copyright © 2021 Buckels, Bolam, Tay and Matthews. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brya G. Matthews, brya.matthews@auckland.ac.nz