





Abstract
After completing this course, the reader will be able to:
Classify the chondrosarcoma subtypes.
Engage in the diagnostic process of chondrosarcoma.
Evaluate the treatment options for chondrosarcoma.
CME Access and take the CME test online and receive 1 AMA PRA Category 1 Credit™ at CME.TheOncologist.com
This review provides an overview of the histopathology, classification, diagnostic procedures, and therapy of skeletal chondrosarcoma. Chondrosarcomas that arise de novo are primary chondrosarcomas, whereas chondrosarcomas developing superimposed on pre-existing benign cartilage neoplasms such as enchondromas or osteochondromas are referred to as secondary chondrosarcomas. Conventional chondrosarcomas can be categorized according to their location in bone into central, peripheral, and juxtacortical chondrosarcomas. Histological grading is related to prognosis; however, it is also subject to interobserver variability. Rare subtypes of chondrosarcoma, including dedifferentiated, mesenchymal, and clear cell chondrosarcoma, are discussed as well. Magnetic resonance imaging is necessary to delineate the extent of the intraosseous and soft tissue involvement preoperatively. Computed tomography is especially recommended in the pelvis and other flat bones where it may be difficult to discern the pattern of bone destruction and the presence of matrix mineralization. Wide, en-bloc excision is the preferred surgical treatment in intermediate- and high-grade chondrosarcoma. In low-grade chondrosarcoma confined to the bone, extensive intralesional curettage followed by local adjuvant treatment and filling the cavity with bone graft has promising long-term clinical results and satisfactory local control. Chondrosarcomas are relatively radiotherapy resistant; therefore, doses >60 Gy are needed in attempts to achieve local control after incomplete resection. Irradiation with protons or other charged particles seems beneficial in this curative situation. Chemotherapy is only possibly effective in mesenchymal chondrosarcoma, and is of uncertain value in dedifferentiated chondrosarcoma. Potential new systemic treatment targets are being discussed.
Introduction
Chondrosarcomas constitute a heterogeneous group of neoplasms that have the production of cartilage matrix by the tumor cells in common [1]. Chondrosarcoma is the third most common primary malignancy of bone after myeloma and osteosarcoma. The majority of these tumors grow slowly and rarely metastasize, and they have an excellent prognosis after adequate surgery. It is generally believed that, because of their extracellular matrix, low percentage of dividing cells, and poor vascularity, they are relatively chemo- and radiotherapy resistant. Wide surgical excision remains the best available treatment for intermediate- to high-grade tumors. However, a minority of patients present or recur with metastatic disease, and up to 13% of recurrent chondrosarcomas are of a higher grade than the original neoplasm [2, 3]. The clinical challenge is to prevent recurrence and to find better treatment options for unresectable or metastatic disease. The radiological and histopathological classification in relation to the clinical symptoms, medical history, and performance status is important for treatment decisions, which should be made by a team consisting of an experienced pathologist, radiologist, medical oncologist, radiotherapist, and orthopedic surgeon. An overview of the classification, clinical characteristics, and therapeutic options is provided in Table 1. New local and systemic treatment options are discussed. This multidisciplinary review addresses the most relevant issues concerning the diagnosis and treatment of chondrosarcoma and provides the current state of the art.
Overview of clinical characteristics and therapeutic options in all subtypes of chondrosarcoma of bone
Overview of clinical characteristics and therapeutic options in all subtypes of chondrosarcoma of bone
Histopathological Classification of Chondrosarcoma
Conventional Chondrosarcoma
Conventional chondrosarcoma of bone constitutes approximately 85% of all chondrosarcomas. Conventional chondrosarcomas can be categorized according to their location in bone into primary central and secondary peripheral chondrosarcomas. The vast majority (>85%) are primary central chondrosarcomas, designated as such based on their location centrally within the medullar cavity. A minority (up to 15%) of conventional chondrosarcomas develop from the surface of bone, most of them as a result of malignant transformation within the cartilage cap of a pre-existing osteochondroma. These are therefore called secondary peripheral chondrosarcomas [4]. A minority (<1%) occur at the surface of bone, possibly of periosteal origin, and are designated periosteal chondrosarcoma. Their histology is similar to that of conventional chondrosarcoma. Juxtacortical chondrosarcoma should be distinguished from secondary peripheral chondrosarcoma because juxtacortical chondrosarcomas have a better prognosis, which is excellent after adequate local surgery alone [5–7].
Central and peripheral chondrosarcomas are histologically similar, and for both, three different grades are discerned, which is at present the best predictor of clinical behavior [3]. Grade I chondrosarcomas are lowly cellular, with an abundant hyaline cartilage matrix, and rarely metastasize [2]. In contrast, grade III chondrosarcomas are highly cellular, with a mucomyxoid matrix and mitoses, with metastases developing in 70% of patients [3] (Fig. 1). Up to 13% of recurrent chondrosarcomas exhibit a higher grade of malignancy than the original neoplasm [2, 3], suggesting chondrosarcomas may biologically progress. Moreover, recurrence of low-grade chondrosarcoma bears the risk for tumor progression toward a higher grade or even dedifferentiation, with a severe adverse prognosis.
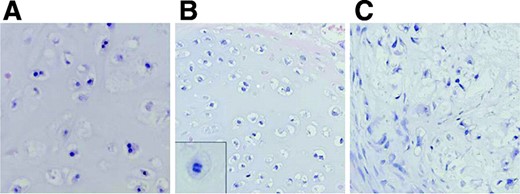
Histology of grade I, II, and III chondrosarcoma. While cellularity is low in grade I chondrosarcoma (A) with chondroid matrix and absent mitoses, in grade II chondrosarcoma (B) mitoses are found (inset). In grade III chondrosarcoma (C), a high cellularity with muco-myxoid matrix changes is seen with cytonuclear atypia (hematoxylin and eosin staining, ∼500×).
The distinction between benign (enchondroma or osteochondroma) and malignant (grade I chondrosarcoma) is, however, extremely difficult both at radiology [8] and histology [9], resulting in high interobserver variability [10]. Fortunately, with new treatment options using curettage and adjuvant phenol application or cryosurgery [11], as described below for enchondroma as well as grade I central chondrosarcoma, the distinction is not always essential for clinical decision making.
Histological grading is, however, also subject to interobserver variability [10]. This is worrisome, because therapy for grade I and grade II chondrosarcoma may differ, as discussed below. Therefore, there is an urgent need for molecular markers independent of histological grade that predict clinical behavior, which may guide clinical decision making. In addition, the elucidation of these markers may also reveal targets for future targeted therapy [12].
Central and peripheral chondrosarcomas differ at the molecular genetic level [13]. The exostosin (EXT) genes, causing multiple osteochondromas (MOs), are involved in the origin of osteochondroma and peripheral chondrosarcoma. In patients suffering hereditary MOs, germline mutations in EXT1 or EXT2 are found [14–16], combined with loss of the remaining wild-type allele in the cartilage cap of the osteochondroma [17]. In contrast, in solitary osteochondromas, EXT1 homozygous deletions are found in the tumor [18]. This results in decreased EXT expression in osteochondroma and secondary peripheral chondrosarcoma [19]. EXT is involved in heparan sulphate biosynthesis [20], and the resulting heparan sulphate proteoglycans (HSPGs) are important for cell signaling. In osteochondromas in which EXT is inactivated, the HSPGs seem to accumulate in the cytoplasm of the cell and Golgi apparatus, instead of being transported to be expressed at the cell surface [19]. This hampers growth signaling pathways, such as the Indian hedgehog (IHH)/parathyroid hormone–like hormone (PTHLH) protein, wingless type (Wnt) protein, and transforming growth factor (TGF)-β signaling pathways that are normally operative during skeletal growth. In peripheral chondrosarcoma, IHH signaling and Wnt signaling are downregulated and TGF-β signaling is upregulated upon increased histological grade. The exact trigger underlying malignant transformation of osteochondroma is unknown. Low-grade peripheral chondrosarcoma is near-haploid [21]. Moreover, PTHLH signaling, which is downstream of IHH and is responsible for chondrocyte proliferation, is absent in osteochondroma, while being upregulated upon malignant transformation [22, 23].
In contrast, in the far more common central chondrosarcoma, EXT is not involved, and the initiating event is still unknown. While enchondromas and low-grade chondrosarcomas are near-diploid and carry few karyotypic abnormalities, high-grade chondrosarcomas are aneuploid and have complex karyotypes [13, 24–26]. Some of the few consistent genetic aberrations include 12q13–15 and 9p21 rearrangements [13, 24, 26–29]. The CDKN2A (p16) tumor suppressor gene, located at 9p21, was shown to be important for chondrosarcoma progression [30, 31]. In addition, the progression from low-grade toward high-grade central chondrosarcoma is characterized by TP53 alterations [25]. Considering the IHH/PTHLH pathway, important in the normal growth plate, PTHLH signaling (PTHLH, parathyroid hormone receptor 1, and B-cell lymphoma 2 protein [BCL2]) was shown to be active in enchondromas as well as chondrosarcomas and increased with increasing histological grade, while IHH signaling was absent [22, 32–35].
Rare Chondrosarcoma Subtypes
In addition to conventional chondrosarcoma, several rare subtypes of chondrosarcoma are discerned, together constituting 10%–15% of all chondrosarcomas (Fig. 2).
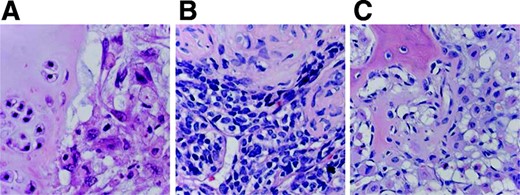
Histology of rare chondrosarcoma subtypes. (A): Histology of dedifferentiated chondrosarcoma with a sharp interface between conventional chondrosarcoma (left) and anaplastic sarcoma (right). (B): Mesenchymal chondrosarcoma with undifferentiated small blue round cells (below) and cartilage differentiation (top). (C): Clear cell chondrosarcoma demonstrating chondrocytes with abundant clear cytoplasm, cartilaginous matrix, and deposition of osteoid. (hematoxylin and eosin staining, ∼500×)
Dedifferentiated chondrosarcoma is a high-grade noncartilaginous sarcoma next to a (usually low-grade) malignant cartilage-forming tumor, with a remarkably sharp junction between the two components [36]. The tumor bears a dismal prognosis [37]. The rare genetic reports on dedifferentiated chondrosarcoma demonstrate that both components share identical genetic aberrations [38, 39], with additional genetic changes in the anaplastic component [38–41], indicating a common precursor cell with early diversion of the two components. No targets for therapy have been reported for dedifferentiated chondrosarcoma.
Mesenchymal chondrosarcoma is a highly malignant lesion that can occur in bone and soft tissue of relatively young patients and is characterized by varying amounts of differentiated cartilage admixed with undifferentiated small round cells [42]. A chromosomal abnormality der(13;21)(q10;q10) was found in two cases [43]. Although 61% of the tumors demonstrate p53 overexpression, no mutations were found [44]. In three cases, expression of the antiapoptotic BCL2, protein kinase C (PKC)-α, and platelet-derived growth factor receptor (PDGFR)-α pathways was found, suggesting targets for treatment, including interferon-α and ciprofloxacin inhibiting PKC-α activity or imatinib inhibiting PDGFR-α signaling [45].
Clear cell chondrosarcoma is a low-grade malignant tumor characterized by tumor cells with clear, empty cytoplasms. Metastases are rare, but may occur up to 24 years after initial diagnosis; thus, long term follow-up is mandatory [46]. A cytogenetic study on four cases suggested that extra copies of chromosome 20 and loss or rearrangements of 9p may be recurrent [47]. Expression of PTHLH, PDGF [48], IHH, Runt related transcription factor 2 [49], and matrix metalloproteinase 2 [50] was demonstrated, suggesting potential targets for future therapy.
It is generally debated whether true myxoid chondrosarcoma of bone exists. Although myxoid chondrosarcoma of bone has similar light microscopic features as extraskeletal myxoid “chondrosarcoma” (EMC), EMC is unrelated to skeletal high-grade (myxoid) chondrosarcoma of bone [51]. The original classification (EMC) as chondrosarcoma was based on positivity for S100, resemblance to chondroblasts on electron microscopy, and focal cartilage formation. Well-formed hyaline cartilage is, however, found in a minority of EMCs [52, 53]. In fact, expression of collagen II and aggrecan, two markers of cartilaginous differentiation, was absent in 86% of EMCs, while S100 expression was very focal or absent [53]. Therefore, the term “chondrosarcoma” in EMC is a misnomer and confusing. The 2002 World Health Organization classification classified this entity in the “tumors of uncertain differentiation” category. The translocation (9;22), which is specific for EMC, is generally absent in myxoid chondrosarcoma of bone and its ultrastructure is different [52]. The few reported cases of bone with proven t(9;22) have a large soft tissue component, which makes the distinction from EMC with bone destruction extremely difficult [54]. Thus, they represent two different entities, and myxoid chondrosarcoma of bone should be regarded as a myxoid variant of intermediate- or high-grade conventional chondrosarcoma.
Radiological Presentation
The distinction between benign and malignant cartilaginous lesions can be difficult. For the distinction between enchondroma and central grade I chondrosarcoma, conventional radiography is not reliable [8, 10, 55, 56]. Localization in the axial skeleton and size >5 cm have been shown to be reliable predictors for malignancy [8]. Dynamic contrast-enhanced magnetic resonance imaging (MRI) shows greater sensitivity [57, 58], although an absolute distinction between benign and malignant cannot be made on radiological grounds alone [8, 59, 60]. Regarding the differential diagnosis of osteochondroma versus low-grade peripheral chondrosarcoma, the thickness and staining characteristics on (dynamic) MRI of the cartilaginous cap provide a rather reliable assessment of the likelihood of malignancy [57].
The locations and radiographic appearances of the different chondrosarcoma subtypes are often characteristic [61], with the mineralized chondroid matrix as a punctate or ring-and-arc pattern of calcifications that may coalesce to form a more radiopaque flocculent pattern of calcification and aggressive features of endosteal scalloping and soft tissue extension. Low-grade chondrosarcoma is shown in Figure 3.
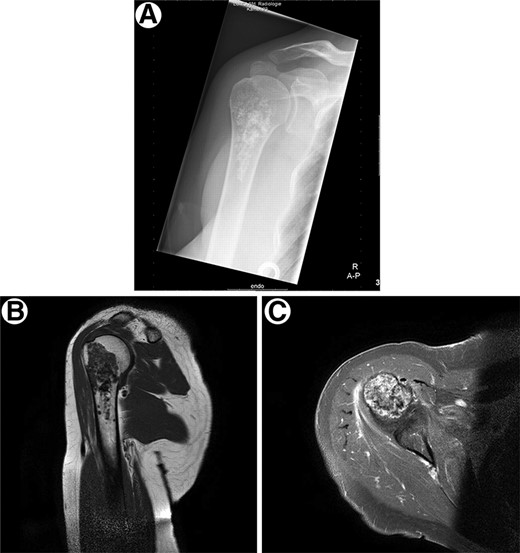
Radiological presentation of a conventional low-grade chondrosarcoma. (A): Conventional low-grade chondrosarcoma in the proximal humerus with chondroid mineralization on conventional radiograph. (B): Discrete endosteal scalloping of the anterior cortex is seen on the T1-weighted magnetic resonance image. (C): Axial T1-weighted magnetic resonance image with fat suppression after i.v. contrast injection demonstrates the typical peripheral ring-and-arc pattern of enhancement. No soft tissue extension is noted.
Aggressive chondrosarcoma subtypes, such as mesenchymal and dedifferentiated chondrosarcomas, often contain areas of matrix mineralization that suggest a chondroid neoplasm; however, they are relatively less extensive than conventional chondrosarcomas, and are usually ill-defined. They may demonstrate intraosseous lytic areas and show aggressive patterns of bone destruction with a moth-eaten to permeate pattern and large soft tissue masses.
Clear cell chondrosarcoma typically has a predilection for the proximal end of the femur and humerus, often with epiphyseal involvement. Radiographs reveal a predominantly lytic lesion. Matrix mineralization is not as frequently apparent in clear cell chondrosarcoma as in conventional chondrosarcoma.
Computed tomography (CT) and MRI are indispensable adjunct tools for optimizing tumor characterization [62]. CT is especially recommended in the pelvis and other flat bones where it may be difficult to discern the pattern of bone destruction and the presence of matrix mineralization. MRI is used to delineate the extent of intraosseous and soft tissue involvement. Evidence of a large unmineralized soft tissue mass associated with a lesion with radiological features indicative of a chondrosarcoma should raise the level of suspicion for dedifferentiation (Figs. 4 and 5).
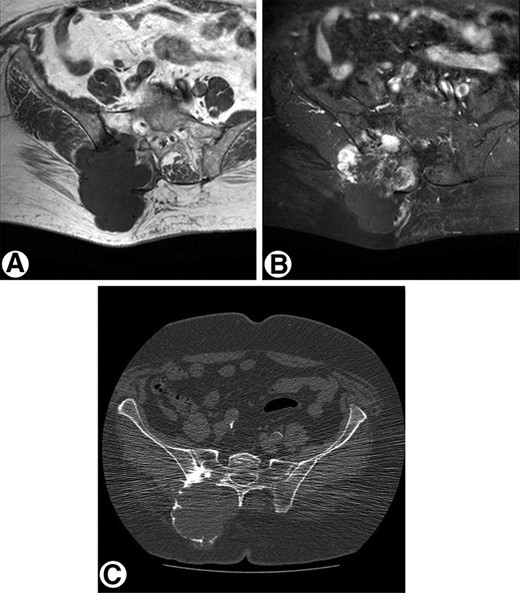
Radiological presentation of a high-grade pelvic chondrosarcoma. (A): Axial T1-weighted magnetic resonance image of high-grade chondrosarcoma arising from the pelvis with cortex destruction and a large soft tissue component. (B): Axial T1-weighted magnetic resonance image with fat suppression after i.v. contrast injection shows areas of solid peripheral enhancement. (C): Computed tomography image demonstrates the bone destruction of the right os ilium and os sacrum with a large soft tissue mass with only peripheral mineralization.
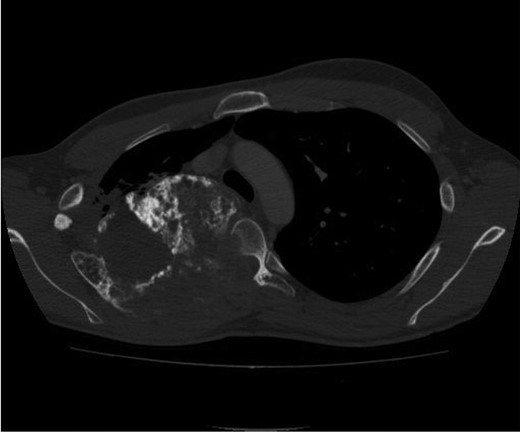
Computed tomography image of mesenchymal chondrosarcoma arising from the posterior chest wall. The large tumor contains extensive areas of matrix mineralization. Aggressive lytic destruction of the posterior part of the rib and the thoracic vertebral body is noted. Histology of these lytic areas shows undifferentiated small round cells.
Surgical Treatment
Although, for all grades and subtypes of nonmetastatic chondrosarcoma, complete surgical treatment offers the only chance for cure, the most optimal type of surgical management is still debated. Wide, en-bloc excision is the preferred surgical treatment of intermediate- and high-grade chondrosarcoma cases [63] (Fig. 6). However, wide excision can lead to considerable morbidity and a demanding reconstruction, depending on the location. On the other hand, in low-grade chondrosarcoma, extensive intralesional curettage followed by local adjuvant treatment, for example, phenolization or cryosurgery (liquid nitrogen), and filling the cavity with bone graft has promising long-term clinical results and satisfactory local control [11, 64]. Local adjuvant treatment can only be successfully used in lesions confined to the bone [64]. However, in some cases of low-grade chondrosarcoma, intralesional excision may not be adequate, for example, because of large size or an intra-articular or pelvic localization. In these cases, wide resection remains the preferable choice for local therapy. Metastases are rare in low-grade chondrosarcoma of the long bones [3]. In general, with current treatment strategies, including local adjuvant therapy, the local recurrence rate is low for grade I chondrosarcoma, but recurrence can occur >10 years postoperatively. In some reports, there were higher local recurrence rates with axial-pelvic tumors of all grades, with a greater tendency to metastasize and/or progress [65], while others were not able to confirm these associations [46, 63]. Local recurrence of a grade I chondrosarcoma in the long bones is associated with worse overall survival, compared with patients without local recurrence [66]. If the local recurrence is solitary, without progression in grade, and located in a long bone, the treatment of choice is an intralesional resection, again with local adjuvant therapy (Fig. 7). In the case of soft tissue involvement in chondrosarcoma grade I, we recommend wide en-bloc resection. Local recurrence of grade II or III chondrosarcoma located in the long and flat bones is treated with a wide excision, although it is often challenging to reach adequate wide resection margins in these patients. Grade I chondrosarcoma in flat bones should also be treated aggressively with a wide resection (Fig. 7).
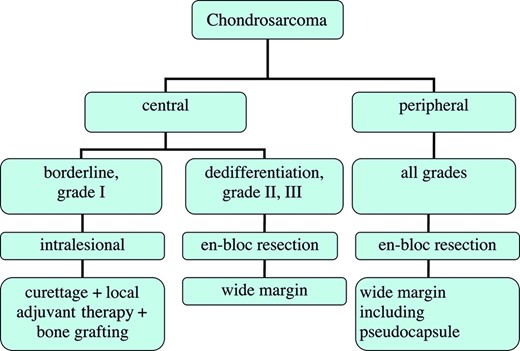
Flowchart of the surgical management of central and peripheral chondrosarcoma.
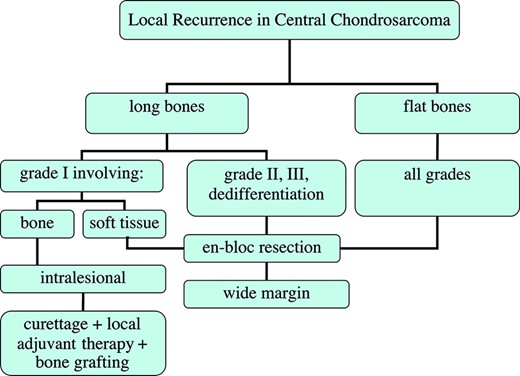
Flowchart of the surgical management of local recurrence in central chondrosarcoma.
In patients with MOs, the development of secondary peripheral chondrosarcoma is rare (assumed in the literature to be <2%) and usually presents after skeletal maturity. Complete surgical removal of the cartilage cap with the pseudocapsule has excellent long-term clinical and local results (Fig. 6). The most common location is the pelvis, where the large cartilage cap can be difficult to excise. In a series of 61 patients with grade I or II secondary peripheral chondrosarcoma of the pelvis published by Donati et al. [67], a 3% local recurrence rate was found after wide resection, in contrast with 23% after inadequate excision.
In our experience, for patients presenting with unresectable lesions of mucoid consistency (usually grade II or III conventional chondrosarcoma), tumor suction is a palliative option for tumor-related pain.
The prognosis for patients with dedifferentiated chondrosarcoma is still poor, despite adequate wide surgical resection and adjuvant systemic therapy. In the case of a pathological fracture of the long bones, wide excision with adequate reconstruction is preferable.
Radiotherapy
As chondrosarcomas grow slowly, with a relatively low fraction of dividing cells, and radiotherapy (RT) acts at dividing cells, chondrogenic tumors are considered relatively RT resistant. RT can be considered in two situations: after incomplete resection, aiming at maximal local control (curative), and in situations where resection is not feasible or would cause unacceptable morbidity (palliative). For curative intentions, doses >60 Gy are required to achieve local control. However, application of this dose with conventional high-energy photon RT is often impossible in the vicinity of critical (neurological) structures, especially in chondrosarcomas arising in the skull base and axial skeleton. Unfortunately, in this situation, postoperative RT is often indicated because these tumors are less accessible for radical resection than lesions in the appendicular skeleton. Given the limitations of conventional RT with photons, alternative radiation modalities have been tested.
Particle therapy with protons has the advantage of a minimal exit dose after energy deposition in the target volume, and hence better sparing of critical structures close to the tumor. Proton RT has been found to be beneficial in incompletely resected chondrogenic tumors of the skull base and axial skeleton. Local control rates of 85%–100% with mixed photon–proton or proton-only protocols (doses up to 79 cobalt Gray equivalents) have been reported by several authors [68–71], with limited severe late effects (<10% incidence of Radiation Therapy Oncology Group [RTOG] grade 3 toxicity). RT with carbon ions or other charged particles represents another attractive radiation modality, which combines the physical advantages of protons with a higher radiobiological activity. Shulz-Ertner et al. [72] recently reported on the effectiveness and toxicity of carbon ion RT in chondrosarcomas of the skull base treated between 1998 and 2005 in Heidelberg. All 54 patients had gross residual disease after surgery. With a median follow-up of 33 months, the actuarial local control rates were 96% and 90% at 3 and 4 years, respectively. RTOG grade 3 late toxicity was lower than with proton RT. Results of RT with helium and neon ions were reported by Castro et al. [73], with a 5-year local control rate of 78%.
Chondrosarcomas that are not resectable and cause complaints might be considered for palliative RT, especially mesenchymal chondrosarcomas, because these tumors are more radiosensitive, in our experience. Limited evidence from older series suggests benefit from conventional RT for chondrosarcomas that cannot be resected. McNaney et al. [74] reviewed the MD Anderson experience with chondrosarcoma in 20 patients. Five of 11 patients treated with RT as monotherapy were controlled with doses of 40–70 Gy; the median follow-up was 53 months. Data on primary RT for chondrosarcomas from Princess Margaret Hospital in Toronto, Canada [75], support the observation that conventional RT can sometimes provide local control and symptom relief.
Systemic Treatment
Chemotherapy is generally not effective in chondrosarcoma, especially in the most frequently observed conventional type and the rare (low-grade) clear cell variant. An additional explanation for this chemotherapy resistance may be expression of the multidrug-resistance 1 gene, P-glycoprotein, resulting in resistance to doxorubicin in vitro [76]. Moreover, the access of anticancer agents may be hampered by the large amount of extracellular matrix and the poor vascularity, implying that the agents have to diffuse over a relatively long distance in order to reach the tumor cells. Moreover, chondrosarcoma grows relatively slowly, while most conventional chemotherapeutic agents act at actively dividing cells. The effect of chemotherapy on grade II and III chondrosarcoma is difficult to assess because the number of reported cases is too low and reported series are mostly retrospective, which may lead to selection bias. There is an urgent need for inclusion of these patients in prospective trials.
Because of a lack of clear evidence, the role of adjuvant chemotherapy in dedifferentiated chondrosarcoma remains unclear, and the standard use of adjuvant chemotherapy outside a clinical protocol should be reconsidered. One study seemed to show an effect on survival [77], which was not confirmed by others [37, 78, 79]. For peripheral dedifferentiated chondrosarcoma, arising in osteochondroma, chemotherapy led to longer survival in a small retrospective report [80]. In a small prospective study of doxorubicin and cisplatin chemotherapy in high-grade spindle cell sarcomas of bone, both patients with dedifferentiated chondrosarcoma had a poor response in their primary tumor, although two of four patients had a complete response of metastases [81].
For mesenchymal chondrosarcoma, there seems to be a role for chemotherapy, although the number of reported cases is even smaller. Huvos et al. [82] published a retrospective series of patients with mesenchymal chondrosarcoma treated with preoperative chemotherapy showing three complete and three partial responses (all T-10 or T-11 protocols) and three nonresponders (all high-dose methotrexate monotherapy) of nine patients treated with preoperative chemotherapy. Nooij et al. [81] prospectively observed one of two good pathological responses in the primary tumor after preoperative doxorubicin–cisplatin combination therapy, but only a limited effect in four patients treated with the same regimen in the metastatic setting (two with stable diseases). Several other publications report cases sensitive to radiochemotherapy [83, 84]. Tumors with a high percentage of small cells and limited cartilage content are thought to be most sensitive to chemotherapy and RT [82], as with other small cell sarcomas. Although prospective studies are lacking, mesenchymal chondrosarcomas are considered sensitive to doxorubicin-based combination chemotherapy as used in other bone tumors. Thus, patients with mesenchymal chondrosarcoma should be considered for adjuvant chemotherapy, and in the case of metastatic disease, palliative chemotherapy.
Additional potential targets for systemic therapy for the different chondrosarcoma subtypes are shown in Table 1 and were extensively reviewed in 2005 [12]. More recently, the antitumor effects of histone deacetylase and aromatase inhibitors were described in chondrosarcoma cell lines, and angiogenesis inhibitors were described in an in vivo model [85–87]. Several phase I studies have reported incidental responses of chondrosarcomas to newer targeted agents, such as vascular endothelial growth factor antisense [88] and recombinant human Apo2L/tumor necrosis factor receptor apoptosis-inducing ligand (TRAIL) [89]. Current phase II trials open for chondrosarcoma patients are using the following drugs: perifosine (serine/threonine kinase Akt inhibitor), apomab (proapoptotic selective agonist of Apo2L/TRAIL death receptor), dasatinib (multitargeted small-molecule tyrosine kinase inhibitor), and the combination of gemcitabine and docetaxel [90]. Also, patients with dedifferentiated chondrosarcomas are allowed in the Euroboss adjuvant trial of the Scandinavian Sarcoma Group [91].
Conclusion
Chondrosarcoma of bone generally has a good prognosis when optimally diagnosed and treated by an experienced team of specialists. Experienced assessment of histological grade and adequate histopathological and radiological (conventional x-ray, MRI, and in some cases CT) classification is used to guide treatment decisions. For conventional low-grade chondrosarcoma confined to the bone, intralesional curettage with local adjuvant therapy (phenol application or cryosurgery) is an option to decrease surgical morbidity. In intermediate- to high-grade tumors, clear margins are necessary to prevent recurrence. It is obvious that recurrences should be avoided, and when they occur they should be treated adequately, especially because they might be of higher histological grade, increasing the risk for metastases and a fatal outcome. Chondrosarcomas are relatively RT resistant; therefore, doses >60 Gy are needed for maximal local control after incomplete resection. Because this high-dose RT is often not feasible with conventional photons, new techniques, such as the use of protons or charged particles, have been tested, with promising results. Chemotherapy is only possibly effective in mesenchymal chondrosarcoma, and is of uncertain value in dedifferentiated chondrosarcoma; both subtypes are rare and bear a poor prognosis. Chemotherapy should preferably be used in clinical trials to define its definite role in chondrosarcoma. Several new drug targets have been identified and phase II studies are currently ongoing. There is an urgent need for new standard systemic treatment options for the minority of patients with unresectable or metastatic disease.
Author Contributions
Conception/design: Hans Gelderblom
Manuscript writing: Hans Gelderblom, Pancras C.W. Hogendoorn, Sander D. Dijkstra, Carla S. van Rijswijk, Augustinus D. Krol, Antonie H.M. Taminiau, Judith V.M.G. Bovée
Final approval of manuscript: Hans Gelderblom, Pancras C.W. Hogendoorn, Sander D. Dijkstra, Carla S. van Rijswijk, Augustinus D. Krol, Antonie H.M. Taminiau, Judith V.M.G. Bovée
References
Available at http://www.clinicaltrials.gov/ct2/results?cond=%22Chondrosarcoma%22. Accessed November 19,
Author notes
Disclosure: No potential conflicts of interest were reported by the authors, planners, reviewers, or staff managers of this article.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}