
Abstract
We describe the effects of recombinant hGH (r-hGH) therapy for up to 6 y on stature and body proportions of 35 children with achondroplasia (Ach). Consecutive height (Ht) measurements were plotted on disease-specific Ach growth curves, but age and sex SD scores (SDS) of Ht, sitting Ht, subischial leg length, and Ht velocity were made with respect to Tanner normal standards. r-hGH was administered by daily subcutaneous injections at a median (range) dose of 30 (15.8–40) U/m2 per week [0.06 (0.04–0.08) mg·kg−1·24 h−1]. Patients were treated for 3 (1–6) y from age 2.25 (1.2–9.3) y. Before treatment, Ht SDS was −4.6 (−6.5 to −3.24). Treatment caused a significant increase in Ht SDS year to year until y 4 (ANOVA F= 46.94;p< 0.01) that was subsequently sustained with no significant further change (y 5 and 6 versus y 4, p> 0.05). When the response to r-hGH was also expressed as a change in Ht velocity, there was a significant increase in the first year of therapy that was maintained over subsequent treatment years (ANOVA = 4.28, p= 0.001). Age was the most important variable accounting for the first-year response in Ht SDS (r2 = 0.41, p< 0.001), and dose of r-hGH did not influence this. Increments in sitting Ht SDS were greater than subischial leg length SDS (F= 26.25, p< 0.001;F= 9.04, p< 0.001, respectively). r-hGH treatment improved the Ht position of Ach children relative to their normal and Ach peers without obvious side effects. A young age at initiation of therapy prevented the characteristic Ht deficit from accumulating. The greater increase in spinal Ht accentuated the existing disproportion. The addition of later surgical leg lengthening could offer the possibility of proportionate adult stature just within the normal range.
Similar content being viewed by others


Main
Ach is the most common genetic form of disproportionately short stature. It is inherited as an autosomal dominant trait and is usually the result of a spontaneous missense mutation in the transmembrane domain of the FGFR3 gene, resulting in a glycine to arginine substitution at codon 380 (1, 2). This mutation constitutively activates the receptor in the absence of ligand (3, 4) and results in the consistent phenotype of Ach, observed worldwide with similar frequency (5). The characteristic phenotype with disproportionately short stature (short limbs), macrocephaly, and the later development of lumbar lordosis and bow legs (6) results in an average adult Ht of approximately 130 (118–145) cm in untreated males and 120 (112–136) cm in females (7).
Linear skeletal growth relies on enchondral ossification of the growth plate cartilage within which chondrocytes undergo a well-defined process of proliferation and maturation (8). The pathogenesis in Ach is a defect in enchondral ossification of bone with normal membranous ossification (9).
GH is an important regulator of linear skeletal growth (10). It directly increases linear growth by stimulating proliferation of epiphyseal growth plate precursor cells, and it also enhances local production of IGF, the latter stimulating clonal expansion of differentiating chondrocytes (11–13). Given the skeletal abnormality, a reduced sensitivity to the action of GH and IGF would be expected (14).
The increased availability of GH with the introduction of r-hGH in 1986 made it possible to test its use in a wider range of conditions than simple replacement therapy for GH deficiency. Its short-term use (1–2 y) in small numbers (n= 6–15) of Ach patients has shown potential benefits of GH in Ach (15–20). Recently, Tanaka et al. (21) also confirmed this in a larger Japanese group (16 to 42 patients treated for a period of 1–3 y).
We describe the effect r-hGH therapy on stature and body proportion, using a wide range of GH doses in a larger number of Ach patients (n= 35) treated over a longer period (1–6 y, median 3 y).
METHODS
We undertook an open study of 35 consecutive children with Ach. The diagnosis was suspected on clinical grounds (22) and confirmed by standard radiologic criteria (6). All patients had cranial and spinal imaging (either computerized tomography or magnetic resonance imaging) after diagnosis with subsequent follow-up scans only if there were clinical concerns (e.g. increased intracranial pressure).
Blood samples for DNA isolation were available from 21 of the 35 patients. DNA was isolated from lymphocytes as previously described (23). The 164-bp product of the transmembrane domain of FGFR3 was amplified from genomic DNA with published oligonucleotide primer sequences (24). Samples were digested with restriction enzyme Sfc 1, Msp I, and Nla III to test for the G1138A, G1138C, and G1138T mutations, respectively.
Anthropometric measurements were taken by accredited measurers during outpatient visits to endocrine clinics at The London Centre for Paediatric Endocrinology. Standard measurements of Ht, SH, and weight were made before treatment and every 6 mo thereafter. Measurements of SILL were derived by calculating the difference between Ht and SH, whereas annual HV was calculated as annualized Ht increments. These measures were converted to age- and sex-appropriate SDS by comparison with normal reference standards (not Ach) as described by Tanner et al. (25). Because Ht is normally distributed, the 50th, 3rd, and 95th percentiles represent the mean and ±2 SD of the normal population. SDS are calculated according to the following formula:EQUATION 1 In this way, statistical comparisons can be accurately made between individuals of different ages and sexes. SILL SDS was not calculated for those <2 y of age, as normal standards are unavailable. Growth was also assessed by plotting serial Ht measurements on disease-specific Ach growth charts (7).

The onset of puberty was defined as testicular volume of at least 4 mL in boys and the onset of breast development in girls and was staged according to Tanner (26). Bone age and Ht predictions were not performed routinely because the abnormal bones prevent interpretation of x-rays in Ach as in other skeletal dysplasias (27).
The dose of GH used in each individual was variable, ranging from physiologic replacement doses of 15–20 U/m2 per week to supraphysiologic doses of >25 U/m2 per week and up to 40 U/m2 per week. The present study is effectively an audit of the long-term results of previous patients randomized to 20 or 40 U/m2 per week for 2 y together with other individuals arbitrarily treated with 30 U/m2 per week and largely reflects personal practice. Those who received <25 U/m2 per week were similar to those receiving >25 U/m2 per week with respect to pretreatment Ht and HV SDS (p> 0.05), and so we were able to correlate response to dose range.
r-hGH (Genotropin, Pharmacia-Upjohn, Stockholm, Sweden or Norditropin, Novo-Nordisk, Denmark) was administered by daily subcutaneous injections at a median dose of 30 (15.8–40) U/m2 per week [i.e. 0.06 (0.04–0.08) mg·kg−1·24 h−1]. Patients were treated for a median (range) of 3 (1–6) y. The number of patients treated year by year were the following: y 1 = 35, y 2 = 27, y 3 = 21, y 4 = 18, y 5 = 9, and y 6 = 6.
Ethical approval was obtained for the present study from the Joint University College London/University College London Hospitals Committee on the Ethics of Human Research. Informed consent was always obtained from parents and assent from the children when appropriate. All patients attended the endocrine clinics at the Middlesex Hospital or Great Ormond Street Hospital for Children, London.
Statistics.
Data are expressed as SDS with respect to Tanner et al. (25) normal values. Plotting raw data of Ht, SH, and SILL on percentiles is an acceptable and easy way of explaining an individual's growth problems to parents. However, in extremely short stature as in Ach in which data lie well below the lowest percentiles, this is not an accurate representation. Further, the statistical analysis of group data requires that the data are presented as a summary statistic with respect to age and sex standardized means, and, in this way, SDS are a well-recognized and robust measure. The SPSS v7.0 statistical package was used for analysis. Examination of data was undertaken to confirm its symmetrical distribution. Our population before treatment was also confirmed to be normally distributed across the Ach-specific growth charts described by Horton et al. (7). Changes in group mean data were compared by 1-way ANOVA with the patients as the blocking variable and Scheffe's post hoc tests for multiple contrasts. Stepwise multiple regression was used to ascertain the effects of age at therapy and dose of GH on the auxologic response, this being expressed as first-year changes in both Ht and HV SDS. Lowess locally weighted regression scatter plot smoothing procedure was used to show the yearly change in HV SDS curves in treated patients and those before treatment (28). This is a robust statistical method that fits a line or curve to include a minimum of 50% of the data points. In this case, it was used to show the inexorable decline in HV seen in Ach patients and as compared with its attenuation in treated Ach (Figure 4). As the data were symmetrically distributed, the mean and median were very similar. We have presented mean and SEM in graphics because this is what is most common, but we thought it important to include median and range to show the wide individual variability of response.

HV SDS changes before and during therapy. Lowess locally weighted regression scatter plot smoothing procedure comparing treated HV SDS data with similar data before treatment and according to age and time. Although the pretreatment value at 11.3 y is shown, it was not included in the analysis of the data, as this case was pubertal at the start of therapy. For three patients, pretreatment HV SDS are not given, as the pretreatment HV was calculated at other centers.
RESULTS
Mutation analysis.
The G1138A mutation in the transmembrane domain of FGFR3 was detected in all 21 patients with Ach from whom DNA was available.
Baseline auxology and puberty status.
The median age at start of therapy was 2.25 (1.2 to 9.3) y. The study included 23 males and 12 females. The median Ht SDS at the start of therapy in the whole group was −4.6 (−6.5 to −3.24), median SH SDS −0.74 (−2.98 to 1.8), and SILL SDS −7.52 (−9.27 to −6.02), confirming the known disproportion.
All were prepubertal at the start of therapy. During the treatment years, the number of patients in puberty were as follows: 2/35 in y 1, 2/27 in y 2, 2/21 in y 3, 3/18 in y 4, 2/9 in y 5, and 2/6 in y 6. Therefore, the majority of our patients remained prepubertal during the period of assessment. These patients will be followed up through puberty until final Ht.
Auxologic response to r-hGH.
The first-year response to r-hGH, expressed as a change in Ht SDS over 1 y of therapy, was greatest in the younger patients (p< 0.05) (stepwise linear regression analysis, R2 = 0.41;p< 0.01).
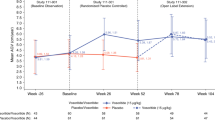
There was a progressive increase in Ht SDS from baseline to y 4 (F= 46.94, p< 0.001) as shown in Figure 1. Years 5 and 6, although different from baseline, were not significantly different from y 4 (p> 0.05, Fig. 1), but the number of cases are small by then. Although it seems that there is no further catch-up in Ht SDS after y 4, it must be remembered that maintaining the SDS position with respect to normal standards is a significant improvement on the expected inexorable decline in Ht SDS seen in Ach and, as such, represents a confirmed benefit. When the raw data are plotted on disease-specific Ach charts, the continued individual improvement becomes evident (Fig. 2).

Change in Ht SDS over time. Serial Ht SDS in 35 Ach children during 6 y of treatment with r-hGH. Data shown as mean ± SEM.

Ht increments over time. Individual linear Ht growth responses of 35 Ach children treated with r-hGH plotted on disease-specific Ach growth percentiles (solid) and shown together with Tanner's normal reference percentiles (dotted) chart (left panel, males;right panel, females). Longitudinal Ht measurements of these patients, plotted on disease-specific growth curves, were continually maintained above the pretreatment percentiles.
We did not observe an obvious dose effect after the first year of treatment with r-hGH. This is shown in Figure 3 and shows the first-year change in Ht SDS with respect to baseline values. All except three patients responded to therapy, but no obvious dose effect was seen. In a small minority, the Ht SDS position was maintained from pretreatment values with no catch-up. However, given the natural history of Ach in which you would expect a decline in Ht position with time, this would be consistent with a positive response.

Dose response. The response to r-hGH therapy in the first treatment year expressed as a change in the first-year Ht SDS in 35 Ach patients does not show a significant dose effect.
The HV of the treated patients before starting r-hGH followed an expected progressive decline. By contrast, treatment significantly and positively changed HV SDS in the first year, and the benefit was sustained in subsequent years (F= 4.28, p< 0.001). GH maintained the growth rates of Ach children close to the mean for normal children, and the slope of the HV SDS decline was significantly less steep than what would be expected from the history of Ach (Fig. 4).
Significant stepwise year-to-year increments were noted in SH SDS from baseline through 3 y (F= 26.25, p< 0.01), but again there were no significant increments thereafter, as shown in the upper panel of Figure 5. The increment in SILL SDS was significant at all years (F= 9.04, p< 0.01), but there were no further increments between treatment years, as shown in the lower panel of Figure 5. In comparison with SH gains, the increase in SILL was minimal, thereby accentuating the existing disproportion (y-4 change in SH SDS versus SILL SDS change, p> 0.05).

Effect on body proportions. Stepwise year-to- year increments in SH SDS (upper panel) and SILL SDS (lower panel) from baseline through 6 y:F= 26.25, p< 0.01;F= 9.04, p< 0.01, respectively. Data shown as mean ± SEM.
DISCUSSION
Since the introduction of r-hGH, the wider indications for GH therapy in short stature conditions not associated with GH insufficiency have attracted increasing interest.
Ach is one of the most severe forms of skeletal dysplasia with individuals on average being 50 cm shorter than the normal adult population. The nature of the growth problem in such conditions is most likely due to reduced sensitivity to the action of GH and IGF (14). Hence, supraphysiologic GH therapy has been used in an attempt to overcome skeletal resistance. Undoubted initial short-term benefits in both Ach and other skeletal dysplasias including Turner's syndrome (15, 16) have been described, although not all have been translated into significant increments in predicted adult Ht (29, 30). Differences in age at onset of therapy, duration and dose of therapy, and accuracy of the method of Ht prediction as well as a secular trend to increased adult Ht (31–33) may have contributed to these discrepancies.
We also report a significant benefit on longitudinal measurements of Ht SDS in an uncontrolled intervention study, but our data were strengthened by a larger number of a mostly prepubertal cohort of much younger age (2.25 y) and longer treatment duration (up to 6 y). In addition, we have been able to assess the effects of a wide range of r-hGH doses on the response. We were not able to show a significant dose response relationship even in the first year of treatment, and this was surprising given the experience in other related conditions (15, 16). However, beyond the first year of life, higher doses have been shown to advance skeletal maturity and to shorten the duration of puberty (34), thereby limiting the eventual growth potential. Thus, it will be important to compare outcomes of low-versus high-dose therapy at adult Ht before making recommendations on the dosage at therapy.
In the natural history of the disease, there is a progressive accumulation of Ht deficit. After the first year of life, the growth rate in untreated children approximates only the third velocity percentile of normal children and remains at this slow rate for the rest of childhood (7). In comparison with the growth rates of normal children, which oscillate about the 50th percentile, this is clearly abnormal. Early r-hGH therapy in children with Ach has prevented the accumulating Ht deficit by maintaining growth velocity near the normal range and maximizing the potential of normal growth in the spine. Although SILL remained significantly compromised, no further deficit was incurred over the treatment period. The accentuation of the existing disproportion in Ach due to the variable SH and SILL responses to r-hGH therapy that was shown in this study has not been reported and, in most cases, has not been examined in previous studies of r-hGH in Ach (15–21). The greater number of patients who had a longer duration of therapy in our study may account for this accentuation of disproportion being shown.
The abnormal bones in Ach make estimates of skeletal maturity unreliable. The inter- and intraobserver error even in the normal situation is well documented (31) and is increased in children with skeletal dysplasias. We have, therefore, not used this as a method of predicting final Ht. Instead, we have used growth charts available from historical Ach patients for comparison (7). Such a method has been used to assess the treatment effects in other conditions (32) and carries with it the potential of misrepresenting final outcomes if there has been a significant secular trend. We showed that our subjects were comparable to historical controls at the time of onset of treatment, whereas the magnitude of the secular trend is unlikely to significantly affect the reported outcome. The magnitude of growth improvement documented was an increase of one Ht SDS over 6 y, an increment of 8 cm in terms of “Ht gained.” Nevertheless, the individual responses reported were variable.
The effects of r-hGH on final Ht are not known, but GH therapy coupled with the opportunity of leg lengthening will alleviate the disproportion and carry the possibility of adult stature for patients within the lower end of the normal range. The dose of r-hGH and the timing(s) of surgical intervention(s) need to be established after adult Ht data are confirmed.
Abbreviations
- Ach:
-
achondroplasia
- FGFR3:
-
fibroblast growth factor receptor 3
- r-hGH:
-
recombinant hGH
- GH:
-
growth hormone
- Ht:
-
height
- SH:
-
sitting height
- SILL:
-
subischial leg length
- HV:
-
height velocity
- SDS:
-
SD scores
References
Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A 1994 Mutations in the gene encoding fibroblast growth factor receptor 3 in achondroplasia. Nature 371: 252–254.
Bellus G, Hefferon TW, Ortiz de Luna R, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano C 1995 Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 56: 368–373.
Webster M, Donoghue DJ 1996 Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J 15: 520–527.
Wilkie AO, Morris-Kay GM, Jones EY, Heath JK 1995 Function of fibroblast growth factors and their receptors. Curr Biol 5: 500–507.
Ikegawa S, Fukushima Y, Isomura M, Takada F, Nakamura Y 1995 Mutations of the fibroblast growth factor receptor-3 gene in one familial and six sporadic cases of achondroplasia in Japanese patients. Hum Genet 96: 309–311.
Wynne-Davies R, Walsh WK, Gormley J 1981 Achondroplasia and hypochondroplasia. J Bone Joint Surg 63: 508–515.
Horton WA, Rotter JI, Rimoin DL, Scott CI, Hall JG 1978 Standard growth curves for achondroplasia. J Pediatr 93: 435–438.
Hunziker EB, Wagner J, Zapf J 1994 Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest 93: 1078–1086.
Rimoin D, Hughes G, Kawfman RL, Rosenthal RE, McAlister WH, Silberberg R 1970 Endochondral ossification in achondroplastic dwarfism. N Engl J Med 283: 728–735.
Isaksson OGP, Lindahl A, Nilsson A, Isgaard J 1988 Action of growth hormone: : current views. Acta Paediatr 343: 12–18.
Daughaday WH, Hall K, Raben MS, Salmon WD Jr, Van den Brande JL, Van Wyk JJ 1972 Somatomedin: : proposed designation factor for sulphation factor. Nature 235: 107
Isaksson OGP, Lindahl A, Nilsson A, Isgaard J 1987 Mechanism of the stimulatory effect of growth hormone on longitudinal bone growth. Endocrin Rev 8: 426–438.
Green H, Morikawa M, Nixon T 1985 A dual effector theory of growth hormone action. Differentiation 29: 195–198.
Zadik Z, Landau H, Chen M, Altman Y, Lieberman E 1992 Assessment of growth hormone (GH) axis in Turner's syndrome using 24-hour integrated concentrations of GH, insulin-like growth factor-I, plasma GH-binding activity, GH binding to IM 9 cells, and GH response to pharmacological stimulation. J Clin Endocrinol Metab 75: 412–416.
Hindmarsh PC, Bridges NA, Brook CGD 1991 Wider indications for treatment with biosynthetic human growth hormone in children. Clin Endocrinol 34: 417–427.
Bridges NA, Brook CGD 1994 Progress report: : growth hormone in skeletal dysplasia. Horm Res 42: 231–234.
Horton WA, Hecht JT, Hood OJ, Marshall RN, Moore WV, Hollowell JG 1992 Growth hormone therapy in achondroplasia. Am J Med Genet 42: 667–670.
Nishi Y, Kajiyama M, Miyagawa S, Fujiwara M, Hamamoto K 1993 Growth hormone therapy in achondroplasia. Acta Endocrinol 128: 394–396.
Weber G, Prinster C, Meneghel M, Russo F, Mora S, Puzzovio M, Del Maschio M, Chiumello G 1996 Human growth hormone treatment in prepubertal children with achondroplasia. Am J Med Genet 61: 396–400.
Shohat M, Tick D, Barakat S, Bu X, Melmed S, Rimoin DL 1996 Short-term recombinant human growth hormone treatment increases growth rate in achondroplasia. J Clin Endocrinol Metab 81: 4033–4037.
Tanaka H, Kubo T, Yamate T, Ono T, Kanzaki S, Seino Y 1998 Effect of growth hormone therapy in children with achondroplasia: : growth pattern, hypothalamic-pituitary function, and genotype. Eur J Endocrinol 138: 275–280.
Oberklaid F, Dances DM, Jennies F, Stacie L, Rosshandler S 1979 Achondroplasia and hypochondroplasia. J Med Genet 16: 140–146.
Ehrlich H 1989 PCR Technologies. Cold Spring Harbour, NY, 35–36.
Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ 1994 Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78: 335–342.
Tanner JM, Whitehouse RH, Takaishi M 1966 Standards from birth to maturity for height, weight, height velocity, and weight velocity: : British children, 1966. Arch Dis Child 41: 613–635.
Tanner JM 1962 Growth at Adolescence. Blackwell Scientific Publications, Oxford
Cox LA 1996 Tanner-Whitehouse method of assessing skeletal maturity: problems and common errors. Horm Res 45: 53–55.
Cleveland WS 1979 Robust locally weighted regression smoothing scatter plots. J Am Stat Assoc 74: 829–836.
Rosenfeld RG, Attie KM, Frane J, Brasel JA, Cara JF, Chernausek S, Gotlin RAW, Chintz J, Lippe BM, Mahoney CP, Moore WV, Saenger P, Johanson AJ 1998 Growth hormone therapy of Turner's syndrome: beneficial effect on adult height. J Pediatr 132: 319–324.
Taback SP, Collu R, Deal CL, Guyda HJ, Salisbury S, Dean HJ 1996 Does growth hormone affect adult height in Turner's syndrome?. Lancet 348: 25–27.
Breunen G, Cameron N 1980 The reproducibility of TW2 skeletal age assessments by a self-taught assessor. Ann Hum Biol 7: 155–162.
Lyon AJ, Preece MA, Grant DB 1985 Growth curve for girls with Turner's syndrome. Arch Dis Child 60: 932–935.
Freeman JV, Cole TJ, Chinn S, Jones PRM, White EM, Preece MA 1995 Cross-sectional stature and weight reference curves for the UK, 1990. Arch Dis Child 73: 17–24.
Darendeliler F, Hindmarsh PC, Brook CG 1990 Dose-response curves for treatment with biosynthetic human growth hormone. J Endocrinol 125: 311–316.
Author information
Authors and Affiliations
Additional information
Supported by Pharmacia-Upjohn, the National Health Service Executive Responsive Funding Programme, and the Children Nationwide Medical Research Fund.
Rights and permissions
About this article
Cite this article
Ramaswami, U., Rumsby, G., Spoudeas, H. et al. Treatment of Achondroplasia with Growth Hormone: Six Years of Experience. Pediatr Res 46, 435 (1999). https://doi.org/10.1203/00006450-199910000-00012
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199910000-00012
This article is cited by
-
Interventions for improving clinical outcomes and health-related quality-of-life for people living with skeletal dysplasias: an evidence gap map
Quality of Life Research (2023)
-
Physical, Mental, and Social Problems of Adolescent and Adult Patients with Achondroplasia
Calcified Tissue International (2019)
-
Final adult height in long-term growth hormone-treated achondroplasia patients
European Journal of Pediatrics (2017)
