


Abstract
Doxorubicin is among the most effective and widely used anticancer drugs in the clinic. However, cardiotoxicity is one of the life-threatening side effects of doxorubicin-based therapy. Dexrazoxane (Zinecard, also known as ICRF-187) has been used in the clinic as a cardioprotectant against doxorubicin cardiotoxicity. The molecular basis for doxorubicin cardiotoxicity and the cardioprotective effect of dexrazoxane, however, is not fully understood. In the present study, we showed that dexrazoxane specifically abolished the DNA damage signal γ-H2AX induced by doxorubicin, but not camptothecin or hydrogen peroxide, in H9C2 cardiomyocytes. Doxorubicin-induced DNA damage was also specifically abolished by the proteasome inhibitors bortezomib and MG132 and much reduced in top2β−/− mouse embryonic fibroblasts (MEF) compared with TOP2β+/+ MEFs, suggesting the involvement of proteasome and DNA topoisomerase IIβ (Top2β). Furthermore, in addition to antagonizing Top2 cleavage complex formation, dexrazoxane also induced rapid degradation of Top2β, which paralleled the reduction of doxorubicin-induced DNA damage. Together, our results suggest that dexrazoxane antagonizes doxorubicin-induced DNA damage through its interference with Top2β, which could implicate Top2β in doxorubicin cardiotoxicity. The specific involvement of proteasome and Top2β in doxorubicin-induced DNA damage is consistent with a model in which proteasomal processing of doxorubicin-induced Top2β-DNA covalent complexes exposes the Top2β-concealed DNA double-strand breaks. [Cancer Res 2007;67(18):8839–46]
Introduction
Doxorubicin (Adriamycin), a topoisomerase II (Top2)-targeting drug, is one of the most effective anticancer drugs used in the clinic. However, doxorubicin-based chemotherapy could result in, among other toxic side effects, life-threatening cardiotoxicity. Patients receiving a cumulative doxorubicin dose of ≥500 mg/m2 have significantly increased risk of developing cardiac toxicity, including cardiomyopathy and congestive heart failure (1–4). Despite the severity of this dose-limiting toxicity, the molecular mechanism underlying doxorubicin cardiotoxicity remains unclear. Currently, the free radical hypothesis is most favored due to the redox cycling ability of doxorubicin (an anthraquinone; refs. 1–4) to generate highly reactive oxygen free radicals.
The cardioprotectant dexrazoxane (Zinecard, also known as ICRF-187) is currently in clinical use to protect against doxorubicin cardiotoxicity (5). The mechanism for the protection has been primarily attributed to iron chelation by the EDTA-like hydrolysis product of dexrazoxane (4), which could decrease the level of hydroxyl free radicals through its chelation of iron (6, 7). However, cardioprotection through iron chelation is still controversial, as the iron chelator ICL670A (deferasirox) shows no protection against doxorubicin despite its efficient iron chelating capability and rapid intracellular distribution (8). Several other free radical scavengers also fail to rescue doxorubicin cardiotoxicity (1).
Dexrazoxane belongs to a class of molecules, bis(2,6-dioxopiperazines), which are known to function as Top2 catalytic inhibitors (9). These compounds are known to antagonize the formation of Top2-DNA covalent (cleavage) complexes through its stabilization of the ATP-bound closed-clamp conformation of Top2 that is unable to access chromosomal DNA (10). In addition, a bis(2,6-dioxopiperazines) derivative, ICRF-193, has been shown to induce degradation of Top2β through a proteasome-dependent pathway (11). It is currently unclear whether the cardioprotective effect of dexrazoxane may involve Top2.
It is well established that the antitumor activity of doxorubicin is due to the formation of a Top2-doxorubicin-DNA ternary complex (the cleavable or cleavage complex; refs. 12–14). There are two Top2 isozymes, Top2α and Top2β, in mammalian cells (15). Doxorubicin, as well as other Top2-directed anticancer drugs such as etoposide (VP-16), amsacrine, and mitoxantrone, targets both isozymes (16, 17). However, the two Top2 isozymes are regulated very differently (18–21). Top2α, which is only expressed in proliferating and tumor cells, plays important roles in cell cycle events, such as DNA replication, chromosome condensation/decondensation, and sister chromatid segregation (15). The high efficacy of doxorubicin chemotherapy is thought to be due to the highly elevated expression of Top2α in cancer cells. By contrast, Top2β is present in all cells, including postmitotic cells (18, 20, 22, 23). Recent studies have suggested that Top2β may play a role in transcription (24, 25). Furthermore, VP-16 has been shown to induce preferential degradation of the Top2β isozyme through a proteasome pathway, which presumably is responsible for the exposure of Top2β-concealed DNA double-strand breaks (DSB; ref. 26). Indeed, recent studies have suggested that the Top2β isozyme is predominantly responsible for the carcinogenic side effect associated with VP-16 chemotherapy (27). However, the role of Top2β in doxorubicin cardiotoxicity is not known. It is noteworthy, however, that Top2β, but not Top2α, is expressed in adult heart (18).
In the present study, we show that dexrazoxane antagonizes doxorubicin-induced DNA damage through its interference with Top2β, which could implicate Top2β in doxorubicin cardiotoxicity.
Materials and Methods
Cell culture and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. TOP2β+/+ and top2β−/− primary mouse embryonic fibroblasts (MEF) were isolated from E13.5 mouse embryos following standard protocols (28). MEFs and H9C2 cardiomyocytes were maintained in DMEM supplemented with 10% FetalPlex animal serum complex (Gemini Bio-Products), l-glutamine (2 mmol/L), penicillin (100 units/mL), and streptomycin (100 μg/mL) in a CO2 (5%) incubator at 37°C. To assay doxorubicin cytotoxicity in MEFs, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was done. MEFs (1.1 × 104) were seeded in 96-well plates for 48 h. Cells were treated with 0.1% DMSO (solvent control) or 200 μmol/L dexrazoxane for 5 h followed by coincubation with doxorubicin for 1 day or VP-16 for 2 days. MTT (0.1 mg) was then added to each well and cells were incubated for an additional 4 h at 37°C. After removal of medium, DMSO was added and absorbance at 570 nm was measured using the Microplate Reader (Bio-Rad). Average IC50 values (mean ± SE) were determined in triplicate or quadruplicate.
Immunoblotting. Cells were lysed in 3× SDS sample buffer [175 mmol/L Tris-HCl (pH 6.8), 15% glycerol, 5% SDS, 300 mmol/L DTT, 0.006% bromphenol blue] followed by boiling for 10 min. Cell lysates were then analyzed by SDS-PAGE. Western blotting was done using anti–γ-H2AX (JBW301, Upstate) and anti–α-tubulin (Developmental Studies Hybridoma Bank) antibodies followed by detection using enhanced chemiluminescence reagents (Pierce). Chemiluminescence signals were then captured using X-ray films or the Kodak Image Station 2000R (for quantification).
Neutral comet assay. Primary MEFs were treated with DMSO or doxorubicin for 1.5 h in a CO2 incubator at 37°C followed by additional 30-min incubation in fresh medium to reverse Top2 cleavage complexes. H9C2 cells were treated with DMSO or dexrazoxane (100 μmol/L) for 3 h, washed, and replenished with fresh medium. Cells were then treated with DMSO or doxorubicin for 1.5 h followed by additional 30-min incubation in fresh medium to reverse Top2 cleavage complexes. Cells were then washed and trypsinized using 0.005% trypsin and resuspended in DMEM supplemented with 10% FetalPlex animal serum complex (10,000/mL). Cell suspension (50 μL) was then mixed with 500 μL 0.5% low-melting point agarose at 37°C. Cell/agarose mixture (75 μL) was transferred onto glass slides. Slides were then immersed in prechilled lysis buffer [2.5 mol/L NaCl, 100 mmol/L EDTA, 10 mmol/L Tris (pH 10.0), 1% Triton X-100, 10% DMSO] for 1 h followed by equilibration in 1× Tris-borate EDTA (TBE) buffer for 30 min. Slides were electrophoresed in 1× TBE at 1.0 V/cm for 10 min and stained with Vistra Green (Amersham Biosciences). Images were visualized under a fluorescence microscope and captured with a charge-coupled device camera. The average comet tail moment was determined from measuring at least 100 cells for each treatment group as described previously (26). Statistical analysis of the mean comet tail moments was done using Student's t test.
Band depletion assay. H9C2 cells (1.2 × 105) were treated with 250 μmol/L VP-16 in the presence or absence of dexrazoxane (150 μmol/L) for 15 min. Cells were either lysed immediately or incubated in drug-free medium for another 30 min at 37°C (to reverse Top2 cleavage complexes) before lysis. Cell lysates were analyzed by Western blotting using the anti-Top2α/Top2β (obtained from Dr. Jaulang Hwang, Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan) and anti–α-tubulin antibody. The amount of Top2 cleavage complexes can be estimated from the difference between the amount of free Top2 after reversal and the amount of free Top2 without reversal.
Homology modeling of the NH2-terminal ATPase domain of human Top2α and Top2β in complex with dexrazoxane. The Modeller (8v2) program was used for construction of the homology models based on the crystal structure of the yeast Top2 ATPase domain in complex with dexrazoxane (ICRF-187; refs. 29–31). The template structure (1QZR.PDB) used to build the model has missing residues and side chains (10). The missing side chains and residues in this structure were replaced and refined using the profix program from the Jackal suite of programs (32–34). The positions of dexrazoxane, ADPNP, and magnesium ions were taken from the yeast crystal structure template (10). The variables for dexrazoxane and the cofactor, ADPNP, were derived from the Amber 9 Antechamber program (35). Partial atomic charges were computed using the AM1-BCC method (36). Each structure model was coarsely refined using the Amber ff03 (37) and general Amber force field with the following energy minimization protocol: 500 steps steepest descents followed by 1,500 steps of conjugate gradient (38, 39).
Results
Dexrazoxane abolishes doxorubicin-induced DNA damage in H9C2 cardiomyocytes. Doxorubicin is known to have two major activities, poisoning of Top2 and induction of reactive oxygen species (ROS) through redox cycling, both of which are known to cause DNA damage. In the current study, we tested whether dexrazoxane could prevent doxorubicin-induced DNA damage in H9C2 cardiomyocytes and determined the molecular mechanism for this effect. As shown in Fig. 1A, doxorubicin induced the DNA damage signal γ-H2AX (Ser139-phosphorylated H2AX, a key DNA damage signal induced by DNA DSBs) in H9C2 cardiomyocytes. Doxorubicin-induced γ-H2AX was concentration dependent up to 1 μmol/L. At higher concentrations of doxorubicin (5 and 10 μmol/L), the γ-H2AX signal was dramatically reduced. This pattern of concentration-dependent inhibition is reminiscent of dose-dependent inhibition of doxorubicin-induced Top2 cleavable/cleavage complexes (12). In the presence of dexrazoxane (200 μmol/L), the doxorubicin-induced γ-H2AX signal was completely blocked. This blocking effect seemed to be specific to Top2-directed drugs, such as doxorubicin and VP-16 (Fig. 1B). γ-H2AX induced by camptothecin (a topoisomerase I poison) and H2O2 in H9C2 cardiomyocytes could not be blocked by cotreatment with dexrazoxane (Fig. 1B).

Dexrazoxane reduces doxorubicin-induced DNA damage. A, 1.5 × 105 H9C2 cardiomyocytes were treated with 0, 0.1, 0.5, 1, 5, and 10 μmol/L of doxorubicin (Doxo) in the presence (+dexrazoxane) or absence (−dexrazoxane) of dexrazoxane (200 μmol/L) for 1 h. Cell lysates were analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody (for assessing protein loading). B, H9C2 cardiomyocytes were treated with 0.1% DMSO (C; for solvent control), 0.1 or 1 μmol/L doxorubicin, 5 μmol/L VP-16 (VP), 10 μmol/L camptothecin (CPT), or 100 μmol/L H2O2 in the presence or absence of dexrazoxane (200 μmol/L) for 1 h. Cells were then lysed and analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody. C, H9C2 cardiomyocytes were treated with 0.1% DMSO (for solvent control), 0.5 μmol/L doxorubicin, or 10 μmol/L VP-16 in the presence (+ICRF-193) or absence (−ICRF-193) of ICRF-193 (50 μmol/L) for 1 h. Cells were then lysed and analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody.
Dexrazoxane reduces doxorubicin-induced DNA damage. A, 1.5 × 105 H9C2 cardiomyocytes were treated with 0, 0.1, 0.5, 1, 5, and 10 μmol/L of doxorubicin (Doxo) in the presence (+dexrazoxane) or absence (−dexrazoxane) of dexrazoxane (200 μmol/L) for 1 h. Cell lysates were analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody (for assessing protein loading). B, H9C2 cardiomyocytes were treated with 0.1% DMSO (C; for solvent control), 0.1 or 1 μmol/L doxorubicin, 5 μmol/L VP-16 (VP), 10 μmol/L camptothecin (CPT), or 100 μmol/L H2O2 in the presence or absence of dexrazoxane (200 μmol/L) for 1 h. Cells were then lysed and analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody. C, H9C2 cardiomyocytes were treated with 0.1% DMSO (for solvent control), 0.5 μmol/L doxorubicin, or 10 μmol/L VP-16 in the presence (+ICRF-193) or absence (−ICRF-193) of ICRF-193 (50 μmol/L) for 1 h. Cells were then lysed and analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody.
To test whether the blocking effect of dexrazoxane was due to inhibition of Top2, another well-characterized Top2 catalytic inhibitor, ICRF-193, was also tested. As shown in Fig. 1C, both the doxorubicin-induced (0.5 μmol/L) and the VP-16–induced (10 μmol/L) DNA damage signal, γ-H2AX, was indeed abolished by cotreatment with ICRF-193 (Fig. 1C).
Doxorubicin-induced DNA damage is blocked by proteasome inhibitors. Doxorubicin-induced DNA damage could be due to either Top2-DNA covalent (cleavable/cleavage) complexes or ROS. As shown in Fig. 2A, doxorubicin-induced γ-H2AX was unaffected by the known ROS scavengers, vitamin C (100 μg/mL) and N-acetylcysteine (100 μg/mL). By contrast, as shown in Fig. 2B, the proteasome inhibitors bortezomib (1 μmol/L) and MG132 (4 μmol/L) significantly reduced (>50% reduction, see Fig. 2B , bottom for quantification) the γ-H2AX signal induced by doxorubicin and VP-16. Recent studies have suggested that proteasomal processing of VP-16–induced Top2-DNA covalent complexes results in the exposure of Top2-concealed DSBs (26). Thus, the involvement of proteasome in doxorubicin-induced γ-H2AX could implicate the involvement of Top2 in doxorubicin-induced DNA damage.

Doxorubicin-induced DNA damage is proteasome dependent. A, H9C2 cardiomyocytes were treated with 0.1% DMSO (for solvent control), 0.5 μmol/L doxorubicin, or 10 μmol/L VP-16 for 1 h in the presence or absence of 100 μg/mL vitamin C (top) or 100 μg/mL N-acetylcysteine (NAC; bottom). Vitamin C and N-acetylcysteine were added 30 min before doxorubicin. Cell lysates were then analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody. B, H9C2 cardiomyocytes were treated with 0.1% DMSO (solvent control), 0.5 μmol/L doxorubicin, or 10 μmol/L VP-16 for 1 h in the presence or absence of either 1 μmol/L bortezomib or 4 μmol/L MG132. Bortezomib and MG132 were added 30 min before doxorubicin or VP-16. Cell lysates were then analyzed by Western blotting (top) using anti–γ-H2AX or anti–α-tubulin antibody. Bottom, quantification of γ-H2AX signals. C, H9C2 cells were treated with 0.1% DMSO, 1 μmol/L bortezomib, or 4 μmol/L MG132 for 30 min followed by cotreatment with either 0.1% DMSO (control) or 0.5 μmol/L doxorubicin for 1.5 h. Neutral comet assay was then done as described in Materials and Methods. The average comet tail moments were plotted as histograms. Columns, mean; bars, SE. *, P < 0.001, t test.
Doxorubicin-induced DNA damage is proteasome dependent. A, H9C2 cardiomyocytes were treated with 0.1% DMSO (for solvent control), 0.5 μmol/L doxorubicin, or 10 μmol/L VP-16 for 1 h in the presence or absence of 100 μg/mL vitamin C (top) or 100 μg/mL N-acetylcysteine (NAC; bottom). Vitamin C and N-acetylcysteine were added 30 min before doxorubicin. Cell lysates were then analyzed by Western blotting using anti–γ-H2AX or anti–α-tubulin antibody. B, H9C2 cardiomyocytes were treated with 0.1% DMSO (solvent control), 0.5 μmol/L doxorubicin, or 10 μmol/L VP-16 for 1 h in the presence or absence of either 1 μmol/L bortezomib or 4 μmol/L MG132. Bortezomib and MG132 were added 30 min before doxorubicin or VP-16. Cell lysates were then analyzed by Western blotting (top) using anti–γ-H2AX or anti–α-tubulin antibody. Bottom, quantification of γ-H2AX signals. C, H9C2 cells were treated with 0.1% DMSO, 1 μmol/L bortezomib, or 4 μmol/L MG132 for 30 min followed by cotreatment with either 0.1% DMSO (control) or 0.5 μmol/L doxorubicin for 1.5 h. Neutral comet assay was then done as described in Materials and Methods. The average comet tail moments were plotted as histograms. Columns, mean; bars, SE. *, P < 0.001, t test.
To test whether DSBs were indeed induced by doxorubicin and prevented by proteasome inhibitors, a neutral comet assay was done. As shown in Fig. 2C, doxorubicin-induced comet tail moment, which reflects the amount of chromosomal DNA DSBs, was significantly reduced by cotreatment with either bortezomib (P < 0.001, t test) or MG132 (P < 0.001, t test). These results further suggest that, similar to VP-16–induced DSBs, doxorubicin-induced DSBs are also Top2 mediated and proteasome dependent.
Doxorubicin-induced DNA damage is Top2β mediated. The Top2β, but not the Top2α, isozyme is expressed in adult heart (18). To test whether Top2β is involved in doxorubicin-induced DNA damage, doxorubicin-induced γ-H2AX was measured in primary MEFs isolated from TOP2β+/+ (wild-type) and top2β−/− (top2β knockout) embryos. As shown in Fig. 3A, doxorubicin-induced γ-H2AX was greatly reduced in top2β−/− MEFs compared with TOP2β+/+ MEFs. Similarly, γ-H2AX induced by VP-16 was also greatly reduced in top2β−/− MEFs (Fig. 3B). By contrast, γ-H2AX was induced by H2O2 and camptothecin to a similar extent in top2β−/− and TOP2β+/+ MEFs (Fig. 3B). These results suggest that the doxorubicin-induced DNA damage signal γ-H2AX is primarily Top2β mediated in MEFs.
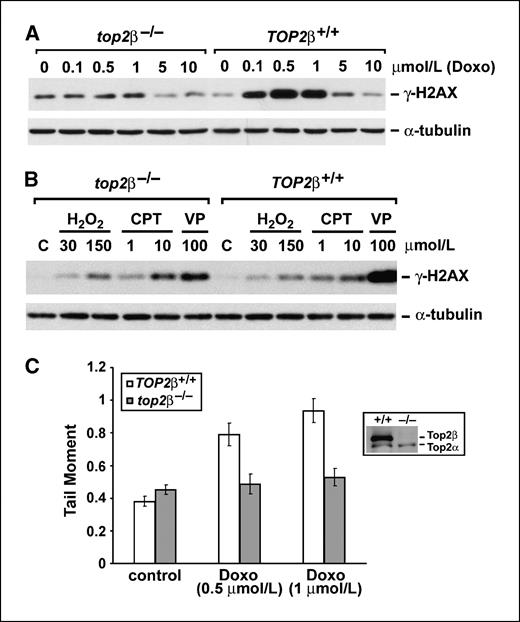
Induction of Top2β-mediated chromosomal DNA DSBs by doxorubicin in primary MEFs. A, 2 × 105 primary top2β−/− and TOP2β+/+ MEFs were treated with 0, 0.1, 0.5, 1, 5, and 10 μmol/L of doxorubicin for 1 h. Cells were lysed and analyzed by Western blotting using either anti–γ-H2AX or anti–α-tubulin antibody. B, primary top2β−/− and TOP2β+/+ MEFs were either untreated (for control) or treated with H2O2 (30 or 150 μmol/L), camptothecin (1 or 10 μmol/L), or VP-16 (100 μmol/L) for 1 h. Cell lysates were analyzed by Western blotting as described in (A). C, primary top2β−/− and TOP2β+/+ MEFs were treated with 0 (control; 0.1% DMSO was added as solvent control), 0.5, or 1 μmol/L of doxorubicin for 1.5 h and neutral comet assay was then done as described in Materials and Methods. Average comet tail moments were plotted as histograms. Columns, mean; bars, SE.
Induction of Top2β-mediated chromosomal DNA DSBs by doxorubicin in primary MEFs. A, 2 × 105 primary top2β−/− and TOP2β+/+ MEFs were treated with 0, 0.1, 0.5, 1, 5, and 10 μmol/L of doxorubicin for 1 h. Cells were lysed and analyzed by Western blotting using either anti–γ-H2AX or anti–α-tubulin antibody. B, primary top2β−/− and TOP2β+/+ MEFs were either untreated (for control) or treated with H2O2 (30 or 150 μmol/L), camptothecin (1 or 10 μmol/L), or VP-16 (100 μmol/L) for 1 h. Cell lysates were analyzed by Western blotting as described in (A). C, primary top2β−/− and TOP2β+/+ MEFs were treated with 0 (control; 0.1% DMSO was added as solvent control), 0.5, or 1 μmol/L of doxorubicin for 1.5 h and neutral comet assay was then done as described in Materials and Methods. Average comet tail moments were plotted as histograms. Columns, mean; bars, SE.
We have also investigated the role of Top2β in doxorubicin-induced chromosomal DNA DSBs using the neutral comet assay. Primary TOP2β+/+ and top2β−/− MEFs were treated with doxorubicin for 1.5 h followed by incubation in drug-free medium for 30 min to reverse doxorubicin-trapped Top2 cleavage complexes. Neutral comet assay was then done to analyze the formation of DSBs. As shown in Fig. 3C, doxorubicin (0.5 and 1 μmol/L) treatment led to a 2-fold increase in comet tail moment compared with control treatment (0.1% DMSO) in TOP2β+/+ MEFs (P < 0.001, t test; white columns), whereas no significant increase (P > 0.2, t test) in comet tail moment was observed in doxorubicin-treated top2β−/− MEFs (black columns). These results suggest that doxorubicin-induced DSBs are primarily Top2β mediated in MEFs.
In addition to doxorubicin-induced DNA damage, we also investigated the role of Top2β and the effect of dexrazoxane on doxorubicin cytotoxicity using MTT assay in confluent (to reduce the contribution from Top2α) primary MEFs. top2β−/− MEFs were shown to be more resistant to doxorubicin than TOP2β+/+ MEFs. The IC50 of doxorubicin in top2β−/− MEFs (2.85 ± 0.10 μmol/L) was significantly higher compared with that in TOP2β+/+ MEFs (0.95 ± 0.06 μmol/L; P < 0.05, t test), suggesting a major role of Top2β in doxorubicin cytotoxicity in primary MEFs. Consistent with this interpretation, top2β−/− MEFs were also shown to be significantly more resistant to VP-16 (another Top2 poison; IC50 = 47.3 ± 4.3 μmol/L) compared with TOP2β+/+ MEFs (IC50 = 28.3 ± 1.0 μmol/L; P < 0.05, t test). As a control, the growth-inhibitory activity of H2O2 was also determined and shown to be the same in top2β−/− MEFs and TOP2β+/+ MEFs (IC50 = 0.2 mmol/L). In addition, dexrazoxane (200 μmol/L) also significantly increased (2- to 3-fold) the IC50 of doxorubicin (2.18 ± 0.16 μmol/L) and VP-16 (95.8 ± 1.9 μmol/L) in TOP2β+/+ MEFs (P < 0.05, t test). By contrast, dexrazoxane had no effect on the growth-inhibitory activity of H2O2 in TOP2β+/+ MEFs (IC50 = 0.2 mmol/L). These results suggest that doxorubicin cytotoxicity is Top2β dependent and dexrazoxane can protect doxorubicin cytotoxicity in primary MEFs.
Dexrazoxane induces proteasomal degradation of Top2β in cardiomyocytes. Our results suggest that Top2β plays an important role in doxorubicin cytotoxicity and doxorubicin-induced DNA damage in primary MEFs. However, it is unclear how dexrazoxane antagonizes doxorubicin cytotoxicity and doxorubicin-induced DNA damage. One possibility is that dexrazoxane antagonizes the formation of doxorubicin-induced Top2 cleavage complexes because dexrazoxane has been shown to reduce Top2 drug-induced protein-DNA cross-links as well as DNA single-strand breaks as monitored by alkaline elution assay (40). Indeed, as shown in Fig. 4A, VP-16 trapped both Top2α (∼70% depleted in free Top2α) and Top2β (∼70% depleted in free Top2β) into Top2-DNA covalent complexes to a similar extent (compare lanes 2 and 3) as evidenced by a band depletion assay. On the other hand, the amount of VP-16–trapped Top2α (∼10% depleted) and Top2β (∼10% depleted) covalent complexes (compare lanes 5 and 6) was much reduced in the presence of dexrazoxane. These results suggest that dexrazoxane can effectively antagonize the formation of VP-16–induced Top2α-DNA and Top2β-DNA covalent (cleavage) complexes in H9C2 cardiomyocytes.

Dexrazoxane induces proteasomal degradation of Top2β in H9C2 cardiomyocytes. A, dexrazoxane antagonizes the formation of Top2α and Top2β-DNA covalent (cleavage) complexes. H9C2 cells were treated with VP-16 in the presence or absence of dexrazoxane (150 μmol/L) for 15 min. The amount of Top2 cleavage complexes was measured by a band depletion assay as described in Materials and Methods. Cells were lysed either immediately or after reversal of the Top2 cleavage complexes (R+250). Cell lysates were analyzed by Western blotting using anti-Top2α/Top2β or anti–α-tubulin antibody. B, 1.2 × 105 H9C2 cells were treated with 100 μmol/L dexrazoxane for indicated times (0, 1, 2, 4, and 6 h). Cells were then lysed and protein levels of Top2α and Top2β isozymes were determined by Western blotting. C, H9C2 cells were treated with 0.1% DMSO (for solvent control), dexrazoxane (100 μmol/L), or ICRF-193 (50 μmol/L) for 2 or 4 h in the presence or absence of the proteasome inhibitor bortezomib (1 μmol/L). Cell lysates were immunoblotted using anti-Top2β antibody. D, H9C2 cells were treated with ICRF187 (100 μmol/L) for 4 h followed by treatment with doxorubicin (0, 0.5, and 1 μmol/L) for 1.5 h. Neutral comet assay was then done as described in Materials and Methods. The average comet tail moments were plotted as histograms. Columns, mean; bars, SE. *, P < 0.001, t test.
Dexrazoxane induces proteasomal degradation of Top2β in H9C2 cardiomyocytes. A, dexrazoxane antagonizes the formation of Top2α and Top2β-DNA covalent (cleavage) complexes. H9C2 cells were treated with VP-16 in the presence or absence of dexrazoxane (150 μmol/L) for 15 min. The amount of Top2 cleavage complexes was measured by a band depletion assay as described in Materials and Methods. Cells were lysed either immediately or after reversal of the Top2 cleavage complexes (R+250). Cell lysates were analyzed by Western blotting using anti-Top2α/Top2β or anti–α-tubulin antibody. B, 1.2 × 105 H9C2 cells were treated with 100 μmol/L dexrazoxane for indicated times (0, 1, 2, 4, and 6 h). Cells were then lysed and protein levels of Top2α and Top2β isozymes were determined by Western blotting. C, H9C2 cells were treated with 0.1% DMSO (for solvent control), dexrazoxane (100 μmol/L), or ICRF-193 (50 μmol/L) for 2 or 4 h in the presence or absence of the proteasome inhibitor bortezomib (1 μmol/L). Cell lysates were immunoblotted using anti-Top2β antibody. D, H9C2 cells were treated with ICRF187 (100 μmol/L) for 4 h followed by treatment with doxorubicin (0, 0.5, and 1 μmol/L) for 1.5 h. Neutral comet assay was then done as described in Materials and Methods. The average comet tail moments were plotted as histograms. Columns, mean; bars, SE. *, P < 0.001, t test.
However, recent studies have also shown that a related compound, ICRF-193, can efficiently induce proteasome-mediated degradation of Top2β (11). Degradation of Top2β is also expected to reduce doxorubicin-induced DNA damage and doxorubicin cytotoxicity in H9C2 cells. We therefore tested the effect of dexrazoxane on the protein level of Top2β in H9C2 cardiomyocytes. As shown in Fig. 4B, treatment of H9C2 cells with 100 μmol/L dexrazoxane induced a time-dependent disappearance of the Top2β isozyme, whereas no significant effect on the level of the Top2α isozyme was observed. Similar to ICRF-193–induced degradation of Top2β, dexrazoxane-induced degradation of Top2β is proteasome mediated. As shown in Fig. 4C, cotreatment of H9C2 cardiomyocytes with the proteasome inhibitor bortezomib abolished dexrazoxane-induced degradation of Top2β. These results suggest that dexrazoxane induces efficient proteasomal degradation of Top2β in H9C2 cardiomyocytes.
To test whether dexrazoxane-induced Top2β degradation could contribute to the protective effect of dexrazoxane on doxorubicin-induced DNA damage, H9C2 cardiomyocytes were pretreated with dexrazoxane for 4 h to induce Top2β degradation and doxorubicin-induced chromosomal DNA DSBs were then measured by the neutral comet assay in the absence of dexrazoxane. As shown in Fig. 4D, dexrazoxane pretreatment effectively reduced doxorubicin-induced comet tail moment (P < 0.001, t test). Together, these results suggest that dexrazoxane could protect doxorubicin-induced DNA damage at least in part through proteasomal degradation of Top2β.
Dexrazoxane targets mammalian Top2α and Top2β isozymes. Our current studies have shown that dexrazoxane can antagonize the formation of both Top2α and Top2β cleavage complexes, suggesting the binding of dexrazoxane to both Top2 isozymes. On the other hand, our current studies have also shown that dexrazoxane induces specific degradation of the Top2β, but not the Top2α, isozyme, which could suggest specific binding of dexrazoxane to Top2β isozyme. To clarify this issue, we did homology modeling studies of hTop2α and hTop2β in complex with dexrazoxane based on the structure of the cocrystal of dexrazoxane and the ATPase domain of yeast Top2 (see Materials and Methods; ref. 10).
As shown in Fig. 5, dexrazoxane was shown to form a tight complex with the ATPase domain of human Top2β at the dimer interface. The overall structure of the human Top2β-dexrazoxane complex is very similar to that of the yeast Top2-dexrazoxane complex (10). In addition, dexrazoxane forms various interactions with the same conserved amino acid side chains (see amino acids at the binding sites in Fig. 5, middle) at the binding site of human Top2β ATPase domain as those of the yeast Top2 ATPase domain. We have also done homology modeling of the human Top2α (ATPase domain)-dexrazoxane complex. The overall structure of the complex is very similar to that of the human Top2β-dexrazoxane complex. Most strikingly, the various interactions between dexrazoxane and the amino acid side chains at the binding sites of the two human isozymes are identical (Fig. 5, middle and bottom). These modeling studies suggest that dexrazoxane can form a tight complex with both human Top2 isozymes.
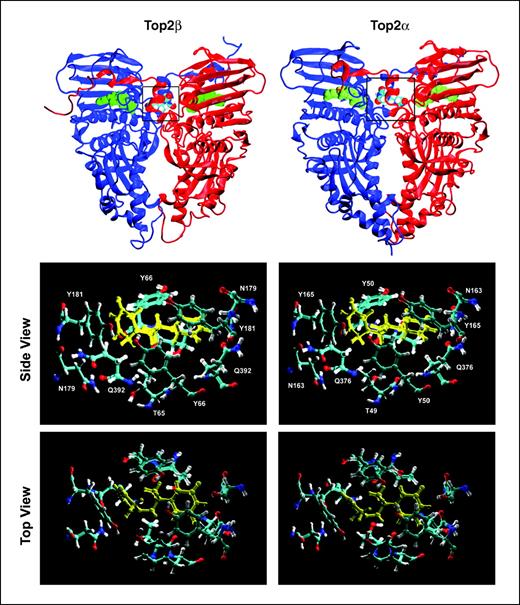
Homology modeling of the NH2-terminal ATPase domain of human Top2α and Top2β in complex with dexrazoxane. Homology modeled structures of the ATPase domain of human Top2β (left) and Top2α (right) in complex with dexrazoxane. Top, the Top2 isozyme dimers are symmetrical with the separate protein chains indicated in red and blue. ADPNP (green) and dexrazoxane (in Corey-Pauling-Koltun coloring) are shown using space-filling models. The dexrazoxane binding region (boxed in top left and top right) is composed of residues from both chains at the dimer interface. The bottom left and bottom right (both side view and top view) show the proximal residues in the dexrazoxane binding sites of human Top2β (left) and Top2α (right) in complex with dexrazoxane (yellow).
Homology modeling of the NH2-terminal ATPase domain of human Top2α and Top2β in complex with dexrazoxane. Homology modeled structures of the ATPase domain of human Top2β (left) and Top2α (right) in complex with dexrazoxane. Top, the Top2 isozyme dimers are symmetrical with the separate protein chains indicated in red and blue. ADPNP (green) and dexrazoxane (in Corey-Pauling-Koltun coloring) are shown using space-filling models. The dexrazoxane binding region (boxed in top left and top right) is composed of residues from both chains at the dimer interface. The bottom left and bottom right (both side view and top view) show the proximal residues in the dexrazoxane binding sites of human Top2β (left) and Top2α (right) in complex with dexrazoxane (yellow).
Discussion
Doxorubicin is one of the most effective and widely used anticancer agents in the clinic. Being an anthraquinone and a strong DNA intercalator, doxorubicin has complex biological activities (41–43). The anticancer activity of doxorubicin has been attributed to its targeting of DNA Top2 by stabilizing a Top2-doxorubicin-DNA covalent complex, referred to as cleavable or cleavage complex (12). This anticancer activity is believed to be related to the ability of doxorubicin to intercalate DNA and hence poisoning of Top2. However, the life-threatening cardiotoxicity associated with the use of doxorubicin is believed to result from the redox cycling activity of doxorubicin due to its quinone moiety (1–4). Both the redox cycling activity, which generates ROS, and the Top2-targeting activity, which generates Top2-DNA covalent complexes, are expected to induce DNA damage. In the current study, we have determined the contribution of these two activities to doxorubicin-induced DNA damage.
We show that doxorubicin induces γ-H2AX, a key DNA damage signal reflecting primarily DNA DSBs, in H9C2 cardiomyocytes. Using this system, we have shown that the doxorubicin-induced DNA damage signal is unlikely to be the result of ROS-mediated DNA damage because vitamin C and N-acetylcysteine cannot attenuate this signal. Instead, several pieces of evidence suggest that the doxorubicin-induced DNA damage signal is primarily due to the formation of Top2-DNA covalent complexes. First, doxorubicin-induced γ-H2AX was shown to be specifically abolished by proteasome inhibitors MG132 and bortezomib. This result is suggestive of an involvement of Top2 because Top2-DNA covalent (cleavage) complexes, unlike other DNA damages (e.g., H2O2-mediated DNA damage), are known to require proteasome for their processing into DNA damage (DSBs; ref. 26). Indeed, doxorubicin is shown to induce chromosomal DNA DSBs in a proteasome-dependent manner (Fig. 2C and see the lower half of the diagram in Fig. 6 for the model). Second, doxorubicin-induced γ-H2AX is much attenuated in top2β−/− MEFs compared with that in TOP2β+/+ MEFs, suggesting the involvement of Top2β. Together, these results suggest the involvement of both Top2-DNA covalent complexes and proteasome in doxorubicin-induced DNA damage, which is consistent with the model that proteasome-mediated degradation of Top2-DNA covalent complexes exposes Top2-concealed DSBs (26).
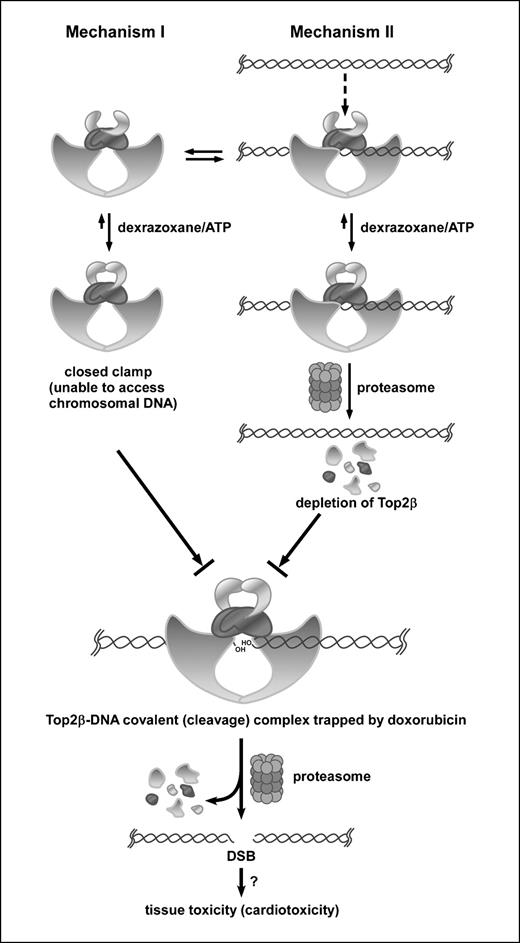
Two proposed mechanisms for the antagonistic effect of dexrazoxane on doxorubicin-induced DNA damage. In this model, only the role of the Top2β isozyme is considered, which would mimic the situation in adult heart where Top2β, but not Top2α, is expressed. Top2β is shown to exist in two states, free Top2β (mechanism I) and DNA-bound Top2β (mechanism II), at equilibrium. Dexrazoxane can bind to Top2β in either state. For mechanism I, binding of dexrazoxane to free Top2β stabilizes the closed-clamp conformation of ATP-bound Top2β and thus prevents binding of Top2β (closed clamp) to chromosomal DNA. Consequently, doxorubicin is unable to trap Top2β into cleavage complexes. For mechanism II, dexrazoxane binds to DNA-bound Top2β and stabilizes the closed-clamp conformation of ATP-bound Top2β, which triggers proteasomal degradation of Top2β (Top2β down-regulation). Top2β down-regulation results in depletion of Top2β and thus fewer doxorubicin-trapped Top2β cleavage complexes. The formation of doxorubicin-trapped Top2β cleavage complexes leads to DNA DSBs through proteasome-mediated processing, which, if not repaired, could contribute to cell death and possible tissue toxicity (e.g., cardiotoxicity).
Two proposed mechanisms for the antagonistic effect of dexrazoxane on doxorubicin-induced DNA damage. In this model, only the role of the Top2β isozyme is considered, which would mimic the situation in adult heart where Top2β, but not Top2α, is expressed. Top2β is shown to exist in two states, free Top2β (mechanism I) and DNA-bound Top2β (mechanism II), at equilibrium. Dexrazoxane can bind to Top2β in either state. For mechanism I, binding of dexrazoxane to free Top2β stabilizes the closed-clamp conformation of ATP-bound Top2β and thus prevents binding of Top2β (closed clamp) to chromosomal DNA. Consequently, doxorubicin is unable to trap Top2β into cleavage complexes. For mechanism II, dexrazoxane binds to DNA-bound Top2β and stabilizes the closed-clamp conformation of ATP-bound Top2β, which triggers proteasomal degradation of Top2β (Top2β down-regulation). Top2β down-regulation results in depletion of Top2β and thus fewer doxorubicin-trapped Top2β cleavage complexes. The formation of doxorubicin-trapped Top2β cleavage complexes leads to DNA DSBs through proteasome-mediated processing, which, if not repaired, could contribute to cell death and possible tissue toxicity (e.g., cardiotoxicity).
We have also shown that dexrazoxane specifically abolished doxorubicin-induced and VP-16–induced, but not camptothecin- and H2O2-induced, γ-H2AX in H9C2 cardiomyocytes. Because both doxorubicin and VP-16, but not camptothecin and H2O2, are Top2 poisons, this result supports the conclusion that dexrazoxane antagonizes doxorubicin-induced DNA damage through its specific interference with Top2. Additional support for this conclusion comes from the use of ICRF-193 (structurally related to dexrazoxane, ICRF-187), which is a well-characterized Top2 catalytic activity inhibitor (9). ICRF-193, which is more potent than dexrazoxane in inhibiting Top2, is shown to be highly effective in antagonizing doxorubicin-induced γ-H2AX in H9C2 cardiomyocytes. The fact that both dexrazoxane and ICRF-193 antagonize the doxorubicin-induced DNA damage signal suggests not only the involvement of Top2 but also a potential mechanism for their antagonism. Bis(2,6-dioxopiperazines), such as ICRF-193 and ICRF-159, are known to stabilize the closed-clamp conformation of ATP-bound Top2 (44, 45). It has been well documented that the closed-clamp conformation of Top2 interferes with the formation of Top2 cleavage complexes induced by Top2-directed drugs possibly due to the inability of the closed-clamp form of Top2 to access chromosomal DNA (46, 47). Consequently, dexrazoxane may antagonize doxorubicin-induced DNA damage through preventing the formation of Top2 cleavage complexes on chromosomal DNA [due to dexrazoxane stabilization of the closed-clamp conformation of Top2 (10), which is unable to access chromosomal DNA].
The identification of Top2β as the major target of doxorubicin to induce DNA damage has suggested a possible new mechanism for the antagonistic effect of dexrazoxane on doxorubicin-induced DNA damage. ICRF-193 is known to induce preferential degradation of the Top2β isozyme through a proteasome pathway, referred to as Top2β down-regulation (11). The reduced Top2β level in ICRF-193–treated cells is expected to decrease the amount of doxorubicin-induced Top2β cleavage complexes and hence reduce DNA damage. Indeed, we have shown that dexrazoxane, like ICRF-193, is highly effective in reducing the level of Top2β (but not Top2α) in H9C2 cardiomyocytes through the activation of a proteasome pathway (Fig. 4). Consequently, dexrazoxane is likely to antagonize doxorubicin-induced DNA damage through two mechanisms: (a) direct interference with the formation of Top2 cleavage complexes and (b) Top2β down-regulation.
At present, it is not clear whether the antagonistic effect of dexrazoxane on doxorubicin-induced DNA damage in H9C2 cardiomyocytes observed in the current study is relevant to the protective effect of dexrazoxane against doxorubicin cardiotoxicity in patients. However, it has been shown that the heart is one of the tissues that prominently express the TOP2β mRNA in adult mice (18). Interestingly, the TOP2α mRNA is completely absent in the heart but still detectable in some other adult tissues, such as the spleen and intestine (18). These findings indicate that Top2β is the only Top2 isozyme that is present in the adult heart and suggest that Top2β targeting by doxorubicin could contribute to its toxic side effects (i.e., cardiotoxicity). In addition, it is known that Top2β can be detected in mitochondria (48) and doxorubicin can accumulate in mitochondria that are abundant in the heart (49). These results suggest that Top2β targeting by doxorubicin in both nuclei and mitochondria of cardiomyocytes could contribute to doxorubicin cardiotoxicity. However, we cannot rule out the involvement of Top2β-independent mechanism(s) (50) for doxorubicin cardiotoxicity, especially at higher doses of doxorubicin, because the DNA damage signal γ-H2AX is significantly reduced at higher concentrations of doxorubicin (Figs. 1A and 3A).
Our current studies, therefore, may have relevance to doxorubicin cardiotoxicity. The two proposed mechanisms (see Fig. 6) for the antagonistic effect of dexrazoxane on doxorubicin-induced DNA damage may have interesting clinical implications. In mechanism I, dexrazoxane stabilizes the closed-clamp form of Top2 and thus prevents access of Top2 to chromosomal DNA. Consequently, doxorubicin is unable to trap Top2 on chromosomal DNA to form Top2-DNA covalent (cleavage) complexes. This mechanism is not Top2 isozyme specific because dexrazoxane can stabilize the closed-clamp forms of both Top2α and Top2β. In fact, our homology modeling studies of the human Top2α and Top2β in complex with dexrazoxane have indicated that the dexrazoxane binding sites are the same for the two isozymes, with identical interactions between dexrazoxane and the various amino acid side chains. There are increasing evidence that the antitumor activity of Top2-targeting drugs is primarily due to Top2α targeting in part due to the overexpression of Top2α in tumor cells. Consequently, dexrazoxane is expected to reduce the antitumor activity of doxorubicin through mechanism I.
By contrast, dexrazoxane can down-regulate the Top2β isozyme specifically through mechanism II (Fig. 6). Through this mechanism, dexrazoxane is expected not to have a major effect on the Top2α isozyme level and hence the antitumor activity of doxorubicin (and other Top2-targeting drugs). If indeed, dexrazoxane, used under the current clinical protocol, prevents doxorubicin cardiotoxicity through both mechanisms, strategies should be developed to prevent mechanism I and favor mechanism II. For example, proper timing of dexrazoxane pretreatment during doxorubicin-based chemotherapy may change the contribution through these two mechanisms.
The idea that Top2 targeting is involved in doxorubicin cardiotoxicity has significant clinical implications. It provides the rationale for developing Top2α-specific anticancer drugs to prevent tissue toxicities (i.e., cardiotoxicity) in patients receiving Top2-based chemotherapy. It is also noteworthy that the involvement of proteasome in Top2β-mediated DNA damage could suggest a novel approach for preventing doxorubicin cardiotoxicity through the combined use of bortezomib (or other proteasome inhibitor) and doxorubicin.
Acknowledgments
Grant support: NIH grant RO1CA102463 (L.F. Liu), Department of Defense Concept Award BC053409 (Y.L. Lyu), and Idea Award BC060915 (Y.L. Lyu).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Dr. Jaulang Hwang for providing the anti-Top2 antibody.