Article Text

Abstract
Patient-reported outcomes (PROs) are used in clinical trials to provide valuable evidence on the impact of disease and treatment on patients’ symptoms, function and quality of life. High-quality PRO data from trials can inform shared decision-making, regulatory and economic analyses and health policy. Recent evidence suggests the PRO content of past trial protocols was often incomplete or unclear, leading to research waste. To address this issue, international, consensus-based, PRO-specific guidelines were developed: the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT)-PRO Extension. The SPIRIT-PRO Extension is a 16-item checklist which aims to improve the content and quality of aspects of clinical trial protocols relating to PRO data collection to minimise research waste, and ultimately better inform patient-centred care. This SPIRIT-PRO explanation and elaboration (E&E) paper provides information to promote understanding and facilitate uptake of the recommended checklist items, including a comprehensive protocol template. For each SPIRIT-PRO item, we provide a detailed description, one or more examples from existing trial protocols and supporting empirical evidence of the item’s importance. We recommend this paper and protocol template be used alongside the SPIRIT 2013 and SPIRIT-PRO Extension paper to optimise the transparent development and review of trial protocols with PROs.
- statistics & research methods
- education & training (see medical education & training)
- protocols & guidelines
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- statistics & research methods
- education & training (see medical education & training)
- protocols & guidelines
- clinical trials
Strengths and limitations of this study
The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT)-patient-reported outcome (PRO) Extension aims to improve the completeness and transparency of trial protocols where PROs are a primary or key secondary outcomes and was developed following Enhancing Quality and Transparency of Health Research Network Guidance.
This explanation and elaboration paper provides information to promote understanding and facilitate uptake of the recommended PRO protocol SPIRIT-PRO checklist items for clinical trials.
A comprehensive protocol template and selected examples from existing trial protocols are provided to facilitate implementation.
The protocol template and explanation and elaboration paper were developed with multistakeholder international input including: trialists, PRO methodologists, psychometricians, patient partners, industry representatives, journal editors, regulators and ethicists.
Although the guidance is limited in focus to clinical trials, many of the SPIRIT-PRO items may also provide useful prompts about PRO content for cohort studies and other non-randomised designs.
Background
Clinical trial protocols are essential documents intended to include the study rationale, intervention, trial design methods, study processes, outcomes, sample size, data collection procedures, proposed analyses and ethical considerations. Provision of sufficient detail is necessary to enable the research team to conduct a high-quality, reproducible study. It also facilities external appraisal of the scientific, methodological and ethical rigour of the trial by relevant stakeholders.1 2 Although trial protocols serve as the foundation for study planning, conduct, reporting and appraisal, they vary greatly in content and quality.1 2 Appraisals of the patient-reported outcome (PRO) content of over 350 past trial protocols revealed that many protocols lack specific information needed for high-quality PRO data collection and evidence generation (online supplemental 1).3–5 As a result, research personnel and potential research participants may not appreciate the purpose of PRO data collection,6 and the need for standardised PRO assessment methods. This may result in high levels of missing data and poor quality or non-reporting of PRO trial results, which may hinder the potential for PRO evidence to be used in regulatory decision-making, health policy and clinical care.6–8 For example, a recent review of cancer portfolio trials illustrates this point; recommended PRO protocol content was frequently not addressed and PRO data from 61 trials, including 49 568 participants, was unpublished.9 Another trial also cited poor PRO completion rates as the reason for not publishing PRO data—and the corresponding trial protocol included only sparse guidance related to the PRO study.7
Supplemental material
In 2013, core protocol guidelines applicable to all types of trials was published based on expert consensus and research evidence, in the form of the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 statement. Its corresponding SPIRIT 2013 explanation and elaboration (E&E) paper provides important information to promote full understanding of, and assist protocol writers to implement, the 33 checklist recommendations.1 2 However, SPIRIT 2013 does not provide specific recommendations about PRO endpoints. PROs can provide valuable information on the risks, benefits and tolerability of an intervention. PRO data are intrinsically subjective, requiring completion by patient-participants within a specific time frame and, as a result, present a range of scientific and logistical challenges for researchers which should be addressed in the trial protocol.6 10–12
To address this issue, international stakeholders worked to develop the SPIRIT-PRO Extension, with the aim of improving PRO content of trial protocols and supporting documents, for use in conjunction with SPIRIT 2013 Guidelines and E&E papers.1 2 The SPIRIT-PRO Extension was published in 2018 and comprises 11 extensions (new, PRO-specific items) and 5 elaborations (an elaboration of an existing SPIRIT 2013 item as applied to clinical trials assessing PROs) recommended for inclusion in clinical trial protocols that have PROs as primary or key secondary outcomes (table 1).10 The SPIRIT-PRO Extension paper reports the 16 items and describes the methods used to develop the checklist, but does not provide detailed implementation instructions or examples. This SPIRIT-PRO E&E paper aims to promote understanding of the guidelines, provide real examples of SPIRIT-PRO items being addressed from a range of different trials and facilitate uptake of the recommended checklist items. A table of contents detailing where to find an example and explanation of each item is provided in table 2. In addition, we describe the development of a new PRO protocol template (online supplemental file 2) for use in protocol development. Additional information and resources regarding the SPIRIT Initiative are available on the SPIRIT website (www.spirit-statement.org).
Supplemental material
SPIRIT 2013 and SPIRIT-PRO Extension checklist: recommended items to address in a clinical trial protocol
Table of contents and resources
The development of the SPIRIT-PRO Extension followed the Enhancing Quality and Transparency of Health Research Network’s methodological framework for guideline development,13 and has been published elsewhere.10 Briefly, these methods included:
a systematic review of existing PRO-specific protocol guidelines to generate the list of potential PRO-specific protocol items;14
refinements to the list and removal of duplicate items by the International Society for Quality of Life Research (ISOQOL) Protocol Checklist Taskforce;
an international stakeholder survey of trial research personnel, PRO methodologists, health economists, psychometricians, patient advocates, funders, industry representatives, journal editors, policy makers, ethicists and researchers responsible for evidence synthesis (distributed by 38 international partner organisations);
an international Delphi exercise;
a consensus meeting in May 2017 to finalise the guidelines and implementation strategy.
International stakeholders provided feedback on the final wording of the SPIRIT-PRO Extension during a final 3-week consultation period. Following minor edits, the guidelines were finalised and agreed by the SPIRIT-PRO Group.10
Development of the PRO protocol template
A PRO protocol template was developed to support implementation of the SPIRIT-PRO guidance (ethical approval ERN_19–0939). The draft template was reviewed by members of the project team and broader SPIRIT-PRO Group, including patient partners. In addition, an international advisory group (IAG), comprising global PRO leads from major pharmaceutical companies, regulators and academics, was convened to review and provide additional feedback on the template. Teleconference meetings were held with members of the SPIRIT-PRO Group and the IAG to discuss the feedback received. Based on the feedback, the template was revised and sent to all for final comments. After a final consultation period, the PRO protocol template was revised and finalised.
Patient and public involvement
Patient partners were involved in the design, conduct, reporting and dissemination plans of our research, including development of the SPIRIT-PRO Extension, the E&E paper, protocol template, tools to support implementation by patient partners and are included as coauthors.15
Glossary
Concept
‘The specific measurement goal (ie, the thing that is to be measured by a PRO instrument). In clinical trials, a PRO instrument can be used to measure the effect of a medical intervention on one or more concepts. PRO concepts represent aspects of how patients function or feel related to a health condition or its treatment’.16
Domain
‘A subconcept represented by a score of an instrument that measures a larger concept comprised multiple domains. For example, psychological function is a larger concept with multiple domains (emotional and cognitive function) that are measured by relevant items.’16
Endpoint*
The variable to be analysed. It is a precisely defined variable intended to reflect an outcome of interest that is statistically analysed to address a particular research question. A precise definition of an endpoint typically specifies the type of assessments made, the timing of those assessments, the assessment tools used and possibly other details, as applicable, such as how multiple assessments within an individual are to be combined17 (eg, change from baseline at 6 weeks in mean fatigue score).18
Health-related quality of life
‘A multidimensional concept that usually includes self-report of the way in which physical, emotional, social or other domains of well-being are affected by a disease or its treatment.’19
Important or key secondary PROs/endpoints
Some PRO measures (particularly health-related quality-of-life measures) are multidimensional, producing several domain-specific outcome scales; for example, pain, fatigue, physical function, psychological distress. For any particular trial, it is likely that a particular PRO or PRO domain(s) will be more relevant than others, reflecting the expected effect(s) of the trial intervention(s) in the target patient population. These relevant PRO(s) and/or domain(s) may additionally constitute the important or key secondary PROs (identified a priori and specified as such in the trial protocol and statistical analysis plan) and will be the focus of hypothesis testing. In a regulatory environment, these outcomes may support a labelling claim. Because these outcomes are linked with hypotheses (Consolidated Standards of Reporting Trials (CONSORT)-PRO Extension 2b),19 they may be subject to p value adjustment (or ‘α spending’). Beyond efficacy/effectiveness, PROs may also be used to capture and provide evidence of safety and tolerability (eg, using the Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events).20
Instrument
‘A means to capture data (eg, a questionnaire) plus all the information and documentation that supports its use. Generally, that includes clearly defined methods and instructions for administration or responding, a standard format for data collection and well-documented methods for scoring, analysis and interpretation of results in the target patient population.’16
Intervention/Treatment
A process or action that is the focus of a clinical study. Interventions include drugs, medical devices, procedures, vaccines and other products that are either investigational or already available. Interventions can also include non-invasive approaches, such as education or modifying diet and exercise.21
Item
‘An individual question, statement or task (and its standardised response options) that is evaluated by the patient to address a particular concept.’16
Observer-reported outcome
‘A measurement based on a report of observable signs, events or behaviours related to a patient’s health condition by someone other than the patient of a healthcare professional.’22
Outcome*
The variable to be measured. It is the measurable characteristic that is influenced or affected by an individuals’ baseline state or an intervention as in a clinical trial or other exposure17 (eg, a fatigue score).
Patient-reported outcome
A PRO is any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else and may include patient assessments of health status, quality of life or symptoms.16 19 PROs are assessed by self-reported questionnaires, referred to as PRO measures or instruments.17
Primary outcome
The most important outcome in a trial, prespecified in the protocol, providing the most clinically relevant evidence directly related to the primary objective of the trial.
Proxy-reported outcome
‘A measurement based on a report by someone other than the patient reporting as if he or she is the patient.’16
Secondary outcomes
Outcomes prespecified in the protocol to assess additional effects of the intervention; some PROs may be identified as important or key secondary outcomes.
SPIRIT Elaboration item
An elaboration of an existing SPIRIT item as applied to a specific context; in this instance, as applied to clinical trials assessing PROs.
SPIRIT-PRO Extension item
An additional checklist item describing PRO protocol content to address an aspect of PRO assessment that is not adequately covered by SPIRIT, as judged by available evidence and expert opinion.
Time-window
A predefined time frame before and after the protocol-specified PRO assessment timepoint, whereby the result would still be deemed to be clinically relevant.23
*The terms outcome and endpoint are often used interchangeably, although this is not always consistent with the range of definitions available. For the definitions included in this glossary, an endpoint is defined from PRO data (ie, the outcome) by fully specifying four components: measurement variable (eg, fatigue ‘in the past week’ as measured by the QLQ-C30), analysis metric (eg, change in fatigue from baseline, final fatigue value, time to clinically important increase in fatigue (and ‘event’), method of aggregation (eg, median fatigue, proportion of patients with severe fatigue, proportion of patients with clinically important change in fatigue) and timepoint. Note that using these definitions, several endpoints can be defined from the same outcome source data, revealing the distinction and relationship between ‘outcome’ and ‘endpoint’ for PROs.
Purpose and development of the explanation and elaboration paper and PRO protocol template
The SPIRIT-PRO Extension, this E&E and the included PRO protocol template are intended to guide the development of trial protocols for ethical review, where PROs are a primary or key secondary outcome, including single-arm and multi-arm trials. We recommend that authors also consider inclusion of checklist items when PROs are exploratory in nature, as appropriate. Protocols may be formatted in accordance with local requirements, however they need to address the SPIRIT-PRO items completely and transparently. The examples provided in this E&E document and protocol template are not intended to be prescriptive about how information is included in protocols, nor how trials be conducted. Trialists may, for example, wish to include a PRO-specific, dedicated section in the protocol with content informed by the SPIRIT-PRO checklist, while others may wish to add PRO content to existing sections of the protocol.
Modelled after other reporting guidelines,2 24 25 this E&E paper presents each checklist item with at least one example from a trial protocol, followed by an explanation of the rationale and main issues to address, to facilitate understanding and usage. The guidelines are intended to be used in conjunction with the SPIRIT-PRO Extension, SPIRIT 2013 Statement and E&E paper and other relevant extensions.1 2 10 26 Empirical data and references to support each SPIRIT-PRO item are provided. Real-world examples for each SPIRIT-PRO item, quoted verbatim, are presented to reflect how key elements could be appropriately described in a trial protocol. These examples were obtained from E&E paper authors, public websites, journals, trial investigators and industry sponsors. Some examples illustrate a specific component of a checklist item, while others encompass all key recommendations for an item. Reference numbers cited in the original quoted text are denoted by (Reference) to distinguish them from references cited in this E&E paper. Health-related quality of life (HRQL) has been used consistently to replace terms for quality of life in examples.
Administrative information
SPIRIT-5a-PRO Elaboration
Specify the individual(s) responsible for the PRO content of the trial protocol.
Example
Trial name: multicentre randomized controlled trial of conventional versus laparoscopic surgery for colorectal cancer within an enhanced recovery programme (EnROL)
PRO endpoints: 1°, 2°
EnROL Trial Management Group
Chief Investigator/Clinical Coordinator
RK, [address, telephone]
Co-Investigator
DK, [address, telephone]
Deputy Clinical Coordinator
TR, [address, telephone]
Trial Management/QA
SP, [address, telephone]; JB, [address, telephone], LD, [address, telephone]; AF, [address, telephone]
Nurse Advisor
SB, [address, telephone]
Statistics
SD, [address, telephone]
Health Economics
PF, [address, telephone]
Quality of Life
JMB, [address, telephone]
Translational Science Advisor
PQ, [address, telephone]
Collaborating Surgeons
HW, [address, telephone]; MG, [address, telephone].27
Explanation
For trials assessing PROs, input from a person with expertise in PRO methodology early in the development phase of the protocol will improve its completeness and quality.10 Providing names and contact details of those contributing to the PRO-specific aspects of the protocol provides recognition, accountability and transparency. It aids identification of competing interests and prevents ghost authorship. It also provides a named point of contact to resolve any PRO-specific queries from other research team members, protocol reviewers and sites (during trial start-up and conduct). Acknowledgements of PRO protocol input from patient-partners as per guidelines for the reporting of patient and public involvement is also recommended.28 Patient and public involvement in all aspects of trial design, including but not limited to: selection of outcomes and measures, timepoints, mode of assessment and reporting, can help minimise burden and ensure that data collected is patient-centred and relevant to participants and to the future patients who will benefit from the research.
Only 7 of 75 (9%) protocols that included PROs from the UK National Institute for Health Research (NIHR) Health Technology Assessment programme explicitly described who was responsible for the PRO component (online supplemental 1).3
SPIRIT-6a-PRO Extension
Describe the PRO-specific research question and rationale for PRO assessment and summarise PRO findings in relevant studies.
Example
Trial name: a phase III, randomised, double-blind, placebo-controlled study evaluating the efficacy and safety of idelalisib (GS-1101) in combination with rituximab for previously treated chronic lymphocytic leukaemia (CLL)
PRO endpoints: 2°
Endpoint selection rationale
Health-related quality of life
‘Direct patient reporting of outcomes using standardised methods has become an increasingly important component of therapeutic assessment. Evaluation of PROs is particularly relevant in patients who cannot be cured of disease (Reference).
PRO questionnaires have been previously used in CLL to understand how patients differ from the general population in terms of health concerns (References), to understand differences in perceptions of well-being in younger versus older patients (References), to determine how treatment affects HRQL (References), and to assess the pharmacoeconomic cost of improvements in HRQL (Reference).
Patients with CLL have overtly impaired well-being relative to comparable controls (References). Fatigue is cited as a common complaint, being present in the substantial majority of patients. Impairment of HRQL prior to any treatment is apparent in those with B symptoms or in patients with anaemia, supporting the concept of initiating treatment when patients experience symptomatic disease. Factors associated with lower overall HRQL have included older age, greater fatigue, severity of comorbid health conditions, advanced stage and ongoing treatment for CLL (Reference). Younger patients appear to have worse emotional and social well-being but older patients experience worse physical HRQL (Reference). In comparative evaluation of chemotherapy-containing regimens, differences in HRQL between therapies (eg, fludarabine vs fludarabine-cyclophosphamide vs chlorambucil) reflected differences in toxicity while greater efficacy was associated with improved HRQL (References).
In this phase III study of GS-1101 and rituximab, it is postulated that incremental GS-1101-mediated tumor control will be correlated with greater positive changes in HRQL and that assessments of the drug’s safety profile will be supported by HRQL evaluations.’29
Explanation
A summary of available PRO evidence and a clearly defined PRO question is required in the background section of the protocol, or a dedicated PRO section if appropriate. Researchers should demonstrate the need for the research and identify the PRO-specific research question to demonstrate the scientific approach and integrity of the PRO study. This should include a review of existing PRO evidence from relevant trials and observational studies (eg, same/similar target population or intervention). This will avoid duplication of research, establish the burden of disease from the patient perspective, identify likely effects of treatment and inform objectives, hypotheses, selection of measures, endpoint definition and analyses (covered by subsequent SPIRIT-PRO items).
Many protocols include PROs without specifying the PRO-specific research question and without a rationale or any reference to PROs in related studies.3 4 9
Provision of this information can inform and motivate research personnel to take note of PRO assessment methods and adhere to standardisation of PRO assessment (eg, when, where, how and who of PRO assessment, as outlined in the protocol under subsequent SPIRIT-PRO items).6 11 Staff who understand the importance of PROs in a trial are able to share this understanding with participants. The combined effect of motivated and co-operative staff and participants may help reduce missing PRO data rates.30 This information is also relevant to research ethics committees/institutional review boards (IRBs) and funders responsible for reviewing the scientific integrity and ethical aspects of the trial.
SPIRIT-7-PRO Extension
State-specific PRO objectives or hypotheses (including relevant PRO concepts/domains).
Examples
Trial group name: Trans Tasman Radiation Oncology Group (TROG)
Trial name: a randomised phase III trial of high-dose palliative radiotherapy (HDPRT) versus concurrent chemotherapy+HDPRT (C-HDPRT) in patients with good performance status, locally advanced or metastatic non-small cell lung cancer (NSCLC) with symptoms predominantly due to intrathoracic disease who are not suitable for radical chemo-radiotherapy (TROG 11.03 P-LUNG GP)
PRO endpoints: 1°, 2°
Objectives
‘The primary objective is to compare, in this group of patients, HDPRT versus C-HDPRT, with respect to
the relief of dyspnoea, cough, haemoptysis and chest pain as assessed by change in total symptom burden from baseline to 6 weeks after the completion of treatment;
response for each component symptom separately (dyspnoea, cough, haemoptysis, chest pain).
The secondary objectives are to compare the two regimens in terms of dysphagia during treatment, thoracic symptom response rate, duration of thoracic symptom response, HRQL, toxicity, progression-free survival (PFS) and overall survival.
The exploratory/tertiary objectives are
to determine how much improvement in HRQL and symptom palliation would be necessary to make the inconvenience due to the longer duration of radiotherapy of C-HDPRT worthwhile, relative to HDPRT. This objective will be addressed in the patient preferences substudy;
to analyse serum protein glycosylation changes and exosomes to identify potential biomarkers of disease response and progression. Prospectively collect and bank tumour tissue and blood samples from this cohort of patients for future evaluation of potential biological markers.’31
Trial name: evaluating different rate control therapies in permanent atrial fibrillation: a prospective, randomised, open-label, blinded endpoint trial comparing digoxin and beta-blockers as initial RAte control Therapy Evaluation in permanent Atrial Fibrillation (RATE-AF)
PRO endpoints: 1°, 2°, exploratory
Hypothesis
‘Null hypothesis for primary outcome: no difference in patient-reported quality of life (measured using the physical functioning domain of the 36-Item Short Form Survey (SF-36) questionnaire) when comparing a strategy of digoxin versus beta-blocker therapy for initial rate control in patients with permanent AF.
Alternative hypothesis: use of digoxin or beta-blocker therapy as initial rate control in patients with permanent AF is superior based on patient-reported quality of life (measured using the physical functioning domain of the SF-36 questionnaire).
Primary objective
Patient-reported quality of life (HRQL), with a predefined focus on physical well-being using the SF-36 physical component summary at 6 months.
Secondary objectives
Generic and AF-specific patient-reported HRQL using the SF-36 global and domain-specific scores, the Atrial Fibrillation Effect on QualiTy of Life (AFEQT) overall score and the 5-level EQ-5D version (EQ-5D-5L) summary index and visual analogue scale at 6 and 12 months.
Echocardiographic left ventricular ejection fraction and diastolic function (E/e’ and composite of diastolic indices) at 12 months.
Functional assessment, including 6-min walking distance achieved, change in European Heart Rhythm Association class and cognitive function at 6 and 12 months.
Change in B-type natriuretic peptide levels as a surrogate for total cardiac strain at 6 months.
Change in heart rate from baseline and group comparison using 24-hour ambulatory ECG.’32
Explanation
The PRO objectives should reflect the research question to be addressed in the trial (SPIRIT-6a-PRO Extension) and be described in the context of the population, intervention, comparator, outcome and timepoint and the estimand framework.33 Study objectives may focus on measuring treatment benefit (superiority), non-inferiority, equivalence. Alternatively, or in addition to one of these objectives, the trial may focus on assessing the safety and tolerability from the patient perspective, or may be more exploratory in nature, where results are presented but no comparative conclusions can be drawn. The PRO-specific study objectives need to clearly align with the proposed analyses methods (SPIRIT-20a-PRO Elaboration). Critically, as described in work by the Setting International Standards in Analysing Patient-Reported Outcomes and Quality of Life Endpoints (SISAQOL) Consortium,34 four key attributes need to be considered a priori for each PRO domain:
Broad PRO research objective/research question.
Between-group PRO objective.
Within-treatment group PRO assumption for the treatment or control arm.
Within-patient/within-treatment PRO objective (please note this component of the objective directly addresses the SPIRIT-12-PRO Extension).
More detailed information on how these can be applied are described in the SISAQOL consensus recommendations.34 Although the SISAQOL recommendations were published for oncology trials, the principles apply more broadly. Prespecification of objectives and hypotheses encourages identification of key PRO domains and timepoints. This is particularly important because PRO data are multidimensional in two important ways. First, there is often more than one relevant PRO in a trial, particularly when the high-level outcome of interest is HRQL. Many HRQL questionnaires yield separate scores for distinct dimensions, such as physical, emotional and social functioning, as well as key symptoms such as fatigue and pain. Second, PRO assessments are typically scheduled at several timepoints during a trial, such as baseline, end of treatment, then a series of long-term follow-ups. Prespecification of objectives and hypotheses—focussing on the most important PRO domains and timepoints—is a good way to reduce multiple statistical testing and avoid selective reporting of PROs based on statistically significant results. Exploratory, hypothesis generating, analyses can also be undertaken but should be specified as such in the final trial report.19 This links to the SPIRIT-20a-PRO Elaboration, which includes plans for addressing multiplicity/type 1 (α) error. The objectives are generally phrased using neutral wording (eg, ‘to compare the effect of treatment A vs treatment B on fatigue’) rather than in terms of a particular direction of effect.2 35 In contrast, the PRO hypothesis states the predicted effect of the interventions on the trial outcomes (eg, ‘patients allocated to treatment A will have less fatigue than those allocated to treatment B’).36–38
Despite the importance of clearly defined PRO objectives and hypotheses, a review of trial protocols determined that 23% failed to include PRO-specific objectives and 81% were missing a clear PRO hypothesis.3
Methods: participants, interventions and outcomes
SPIRIT-10-PRO Extension
Specify any PRO-specific eligibility criteria (eg, language/reading requirements or prerandomisation completion of PRO).30 If PROs will not be collected from the entire study sample, provide a rationale and describe the method for obtaining the PRO subsample.
Example
Trial group name: South West Oncology Group (SWOG)
Trial name: health status and quality of life in patients with early stage Hodgkin’s disease: a companion study to SWOG-9133 (SWOG S9208)
PRO endpoints: 1°, 2°
‘Eligibility criteria
Ages eligible for study: 18 years and older (adult, older adult)
Sexes eligible for study: All
Accepts healthy volunteers: No
Sampling method: Non-probability sample
Study population: community sample.
Criteria: disease characteristics: patients must be eligible for and registered to SWOG-9133.
Patient characteristics: patients must be able to complete the questionnaires in English. If they are not able to complete questionnaires in English, patients may be registered to SWOG-9133 without participating in SWOG-9208.’
The Symptom and Personal Information Questionnaire #1, the Cancer Rehabilitation Evaluation System Short Form and Cover Sheet must be completed prior to registration and randomisation on SWOG-9133.39
Explanation
Any eligibility criteria relevant to PRO assessment should be considered during the trial design and clearly specified in the protocol for consistent use by research personnel. In some trials, the baseline PRO assessment is required before randomisation as an eligibility criterion.30 This helps to ensure there will be a valid baseline questionnaire from all patients, which is essential for calculation of change scores, or inclusion as a covariate in modelling longitudinal PRO data. For unblinded trials, this also ensures PRO data are collected before participants are aware of the randomisation which may affect some aspects of the participant’s response, for example, anxiety/emotional well-being.40 In the absence of such an eligibility criterion, there is a risk that the baseline assessment may be conducted after randomisation but before the intervention is administered, resulting in detection bias. The maximum time between this assessment and randomisation should be defined and should not be too long.
It may not always be possible to collect PROs from all study participants, for example, due to non-availability of questionnaires in appropriate languages (see SPIRIT-18a(iii)-PRO Extension),9 literacy requirements or due to cognitive function (see SPIRIT-18a(iv)-PRO Extension). These PRO-relevant exclusions typically should not preclude the affected participants from enrolling in the trial, unless the PRO is the primary outcome. Evidence suggests eligibility criteria as stated in trial protocols often differ from what is finally reported in the trial publication,41 and data on use of other language or culturally appropriate PRO instruments is often missing from the protocol.13 42
Where the needs of specific groups have been identified (eg, not fluent in English) but not accommodated in the study protocol (eg, non-English language versions not available, assistance with reading and writing English version not permitted), this should be stated, and the rationale for the sampling method described and justified. Trialists should aim to be as inclusive as possible. Given the significance of PROs, the research community has a moral obligation to, where possible, to address gaps in availability of culturally validated PRO instruments. In the meantime, the implications for generalisability of findings should be discussed in subsequent publications.19
SPIRIT-12-PRO Extension
Specify the PRO concepts/domains used to evaluate the intervention (eg, overall HRQL, specific domain, specific symptom) and, for each one, the analysis metric (eg, change from baseline, final value, time to event) and the principal timepoint or period of interest.
Examples
Trial name: evaluating different rate control therapies in permanent atrial fibrillation: a prospective, randomised, open-label, blinded endpoint trial comparing digoxin and beta-blockers as (RATE-AF)
PRO endpoints: 1°, 2°, exploratory
Primary outcome
‘Patient-reported quality of life (HRQL)—SF-36 physical component summary score at 6 months.
Secondary outcomes
Patient-reported HRQL
SF-36 global and domain-specific scores at 6 and 12 months.
EQ-5D-5L summary index and visual analogue scale at 6 and 12 months.
AFEQT overall score at 6 and 12 months.’32
Trial name: a phase III, randomised, double-blind, placebo-controlled study evaluating the efficacy and safety of idelalisib (GS-1101) in combination with rituximab for previously treated CLL
PRO endpoints: 2°
‘Change in HRQL domain and symptom scores based on the Functional Assessment of Cancer Therapy: Leukaemia (FACT-Leu)—defined as the change from baseline and the time to definitive increments or decrements of 10%, 20% and 40% from baseline; time to definitive increment (better than baseline by the specified amount) is the interval from randomisation to the first timepoint when the HRQL measure is consistently better than at baseline (including that timepoint as well as all the subsequent timepoints) in a subject whose last HRQL score is better than at baseline; and time to definitive HRQL decrement (worse than baseline by the specified amount) is the interval from randomisation to the earliest of death or the first timepoint when the HRQL measure is consistently worse than at baseline (including that timepoint as well as all the subsequent timepoints) in a subject whose last performance status score is worse than at baseline.’29
Explanation
For each outcome, including PROs, the trial protocol should define four components: the specific measurement variable, which corresponds to the data collected directly from trial participants (eg, Beck Depression Inventory score, all-cause mortality); the participant-level analysis metric, which corresponds to the format of the outcome data that will be used from each trial participant for analysis (eg, change from baseline, final value, time to event); the method of aggregation, which refers to the summary measure format for each study group (eg, mean, proportion with score >2) and the specific measurement timepoint of interest for analysis.1 Many PRO questionnaires are multidimensional, assessing multiple facets of the impact of a disease and its treatment and usually include multiple assessments over the course of the trial. The multidimensional nature of PROs is most apparent in HRQL questionnaires, which often include various aspects of functioning and symptoms, which are often scored as distinct ‘domains’. These domains may not be affected equally by the trial interventions. The SPIRIT-7-PRO Extension encourages protocol writers to identify the domains that are most likely to be affected in the trial objectives and hypotheses, drawing on previous evidence (SPIRIT-6a-PRO Extension). The SPIRIT-12-PRO Extension item reinforces the statement of these key domains, and also the most important timepoints (ie, where greatest impact of interventions are expected), and develops that concept further by encouraging protocol contributors to think about how these PRO domains and timepoints will be analysed, that is, the analysis metric.34 To ensure transparency and credibility of the analysis, it is recommended that there is prespecification of the PRO concepts/domains, analysis metric(s) and timepoint(s) of interest, whether the PRO is a primary, secondary or exploratory outcome. These should closely align with the study hypotheses/objectives and the nature and trajectory of the disease or condition under investigation.34 43 The selected key domains, timepoints and analysis metric should be used to specify the PRO endpoints, integrated in the full endpoint model of the trial.
A clearly defined endpoint model, organising all trial outcomes (PRO and non-PRO), typically in primary, secondary and exploratory endpoints, allows rigorous control of the evidence demonstration, especially the control of the statistical testing. Each PRO endpoint in the model should explicitly specify a single domain and a single time horizon. The endpoint model enables procedures for type I error control (risk of false positive finding) (see SPIRIT-20a-PRO Elaboration). Broadly, the concepts and domains (sub concepts) measured by a PRO may be ‘proximal’ in nature, that is, direct impact of the disease and treatment (eg, symptoms such as pain, fatigue, nausea, rash and anxiety) or more distal, ‘knock-on’ effects, (eg, functional status and global quality of life), as illustrated for ovarian cancer in (figure 1, inspired by the Wilson and Cleary model44). Of note, the Food and Drug Administration (FDA) are increasingly focused on the individual measurement of well-defined concepts that impact on HRQL but are more proximal to a therapy’s effect on the patient and the patient’s disease: symptomatic adverse events, physical function and, where appropriate, a measure of the key symptoms of the disease.45

Proximal and distal effects of therapy on patient symptoms and quality of life. Adapted from Wilson and Cleary.44
Common analysis metrics may include magnitude of event at time t, proportion of responders at time t, overall PRO score over time or response patterns/profiles. These should be prespecified alongside the levels of statistical and clinical significance for the study and any responder definition in use.16 Timepoints for analysis should be chosen to best address the research question, while taking into account aspects such as the natural history of the disease/condition and its treatment, the PRO measurement properties and recall period and participant completion burden.16 46
The example, idelalisib and rituximab improve PFS over rituximab alone in unfit patients with relapsed CLL: a phase III study, illustrates a ‘time to event’ PRO endpoint or analysis metric, where the event is definitive improvement or definitive deterioration in a PRO. This approach allows repeated PRO measurements to be converted to a single measure: time to definitive increment or decrement. This requires quite complex and specific criteria for degree and duration of change. Also, this particular example does not specify any key domains of the FACT-Leu, but rather applies this analysis metric to all HRQL domain and symptom scores. In contrast, the RATE-AF example identifies a single score (the SF-36 physical component score), and a specific timepoint (6 months) as the primary outcome, with other SF-36 domains, questionnaires and timepoints specified as secondary outcomes.
SPIRIT-13-PRO Extension
Includes a schedule of PRO assessments, providing a rationale for the timepoints and justifying if the initial assessment is not prerandomisation. Specify time-windows, whether PRO collection is prior to clinical assessments, and, if using multiple questionnaires, whether order of administration will be standardised.
Examples
Trial group: TROG
Trial name: a randomised phase III trial of HDPRT versus concurrent C-HDPRT in patients with good performance status, locally advanced or metastatic NSCLC with symptoms predominantly due to intrathoracic disease who are not suitable for radical chemo-radiotherapy (TROG 11.03 P-LUNG GP) (figure 2).31

Example schedule of patient-reported outcome (PRO) assessments in the Trans Tasman Radiation Oncology Group 11.03 P-Lung GP Trial. QoL, quality of life.
Trial group: UK Medical Research Council Scottish Cancer Trials Breast Group in association with: Breast International Group
Trial name: MRC phase III randomised trial to assess the role of adjuvant chest wall irradiation in ‘intermediate risk’ operable breast cancer following mastectomy (MRC SUPREMO TRIAL (BIG 2–04) (figures 3 and 4))
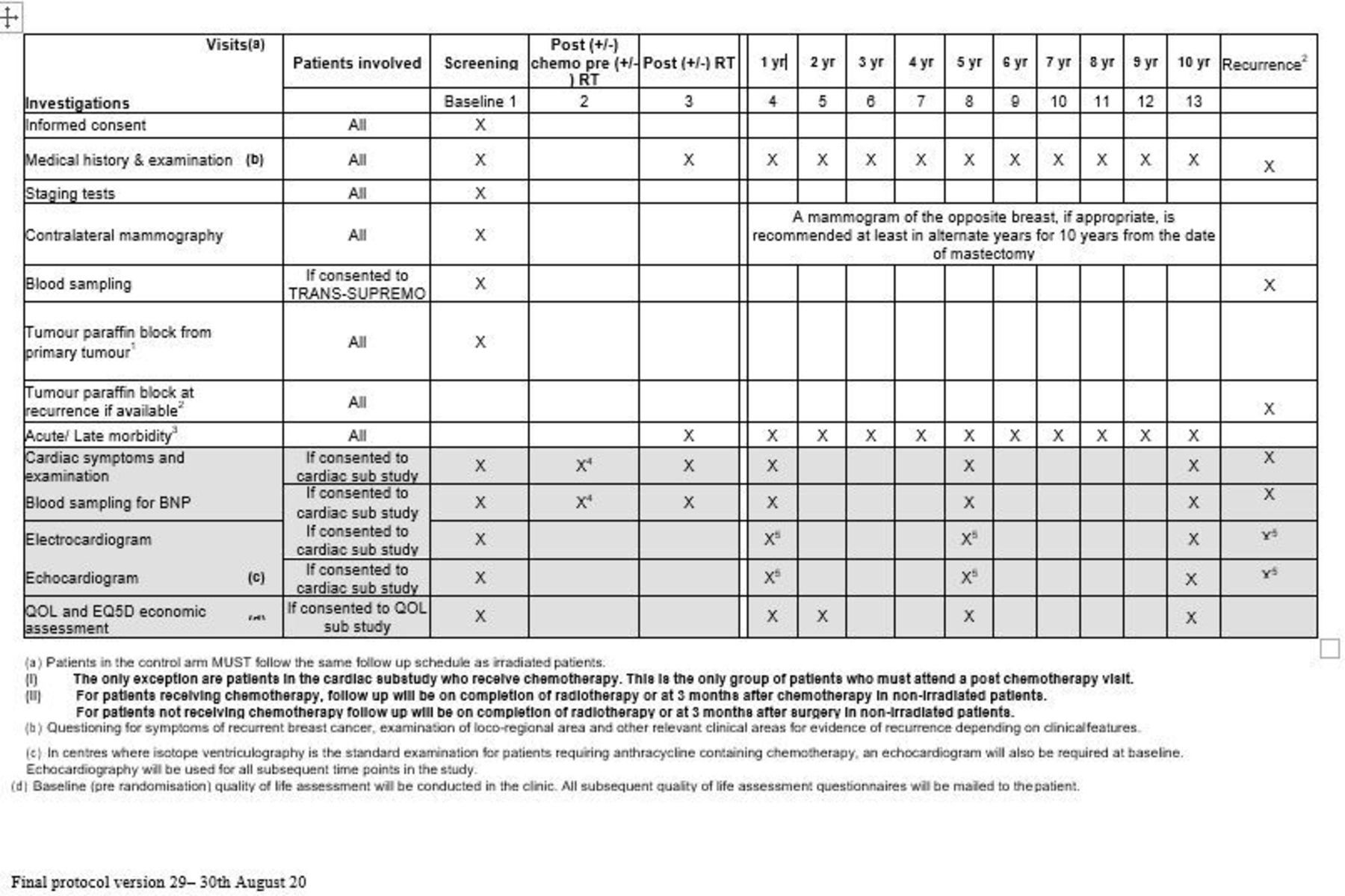
Example schedule of patient-reported outcome assessments in the MRC SUPREMO TRIAL (BIG 2–04).140 BNP, B-type natriuretic peptide; QoL, quality of life.
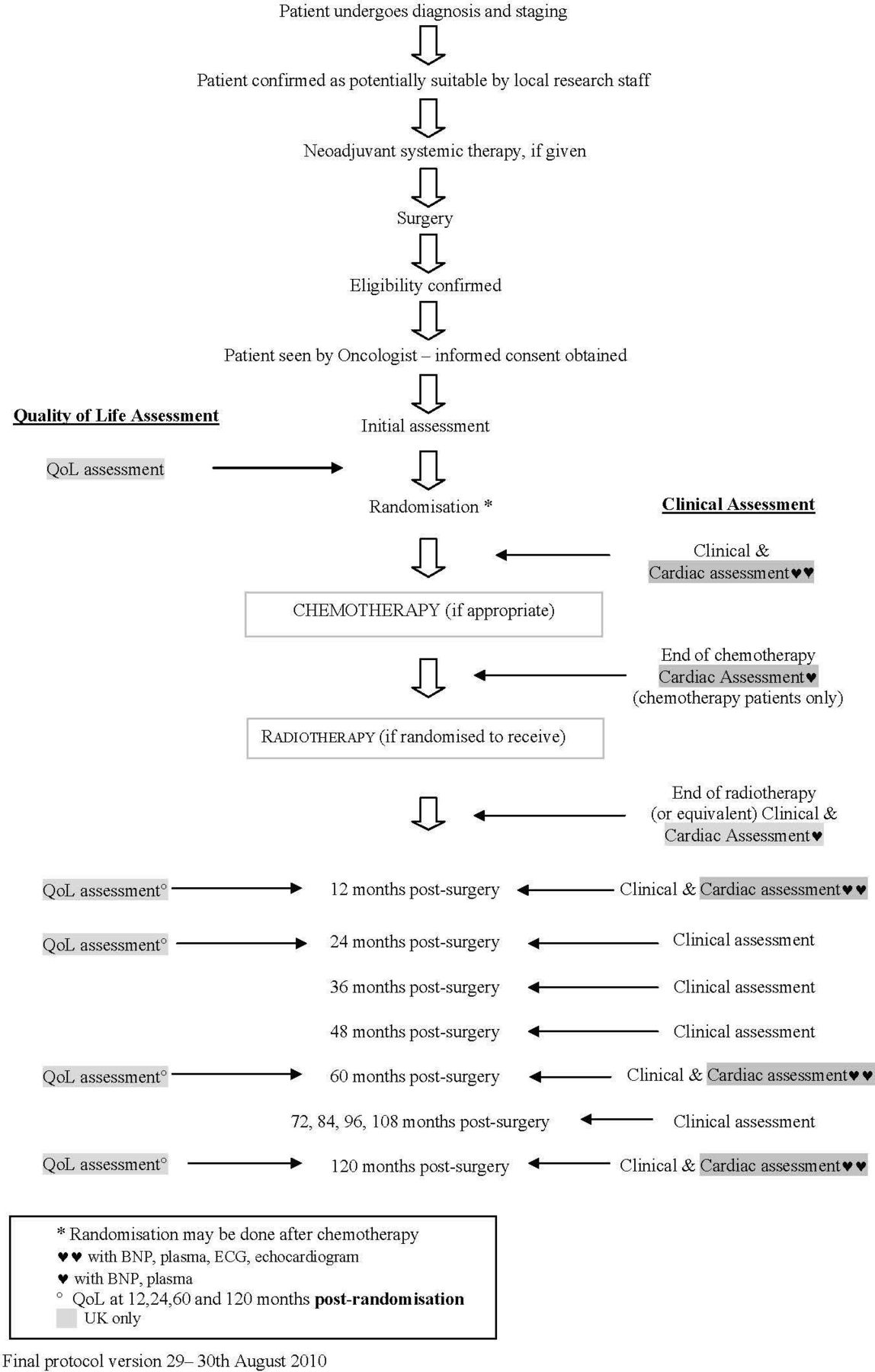
Flow diagram schedule of patient-reported outcome assessments in the MRC SUPREMO TRIAL (BIG 2–04).140 BNP, B-type natriuretic peptide; QoL, quality of life.
Explanation
A clear and concise schedule of PRO assessments (figures 2–4) can: assist trial staff to be organised and prepared for participant visits, inform study participants about the methods and expectations of trial participation and facilitate review of participant burden by research ethics committees/IRBs.30 The scheduled PRO assessments should provide the data required to address the study’s PRO objectives. When selecting appropriate timepoints for assessment, it is important to consider the natural history of disease/progression, the hypothesised impact of therapy over time and practical considerations such as alignment of assessments with clinic visits and recall period of PRO measures. PRO assessments should be described in the protocol text and in the schedule of assessment table along with the other clinical data collection activities, for ease of reference. This is recommended whether the PRO is completed by the participant during study visits or outside of the study visits (eg, at home).
The timing of the baseline PRO assessment relative to other study-related events is important and therefore should be specified in the schedule of assessments. Collecting PRO data prior to randomisation helps ensure an unbiased baseline assessment, and if specified as an eligibility criterion, can promote data completeness (SPIRIT-10-PRO Extension). Baseline PRO data are often used as a covariate in analyses and are essential to calculating change from baseline, however, collecting data from enrolled patients prior to randomisation can be logistically challenging. One approach is to have participants complete the baseline PRO assessment immediately after providing consent, while the site staff obtain the randomisation assignment from the study system. However, there may be scenarios in which prerandomisation PRO assessment is unnecessary or not possible, for example, emergency surgery trials.
Stating the time-windows for each PRO assessment clearly in the protocol text and schedule of assessments table or footnote will help staff adhere to them. Examples of time-windows for PRO assessment are similar to time-windows for other types of assessments, such as a study visit that may occur on day 10–14 postbaseline, or on day 30±3 days postsurgery. Time-windows for each scheduled PRO assessment require an unambiguous reference point, to ensure that PRO data collection captures clinically relevant timepoints of interest. In deciding the size of the time-window for a PRO assessment, consider the trade-off between a smaller, more precise, time-window and a larger more feasible window. One approach is to specify a time-window that is a little larger than the ideal and not allow exceptions; this approach is more consistent than setting a smaller time-window and allowing exceptions. Often PRO assessments that occur during active treatment, for example, chemotherapy, have a smaller time-window to capture acute toxicity that arise and resolve relatively quickly, while those occurring many months or years after treatment completion can have a larger window if the participant’s outcomes are expected to stabilise over time.
When the PRO assessment occurs during a research or clinic visit, it is recommended that PRO assessment is standardised to be completed prior to clinical consultation, assessments or procedures. For example, if a PRO instrument assesses participants’ experiences of pain in the past 7 days, and the study visit includes a bone marrow biopsy, the schedule of assessments should indicate that the PRO assessment be completed prior to the biopsy. This will prevent the pain assessment from capturing pain associated with the biopsy, reduce risk of missing data as participants may not feel well enough to complete PROs following their procedure and offer a ‘routine’ for study staff responsible for data collection.
When more than one PRO questionnaire is scheduled, it is recommended that the order of questionnaires is standardised, with those higher in the endpoint hierarchy being collected first.
These two forms of standardisation of PRO administration are examples of the more general principle in research methodology that standardisation of methods reduces unwanted sources of variation, whether random (ie, no net effect on estimates of interest, such as the impact of interventions on PROs) or systematic (ie, causing bias).
SPIRIT-14-PRO Elaboration
When a PRO is the primary endpoint, state the required sample size (and how it was determined) and recruitment target (accounting for expected loss to follow-up). If sample size is not established based on the PRO endpoint, then discuss the power of the principal PRO analyses.
Examples
Trial name: the chronic autoimmune thyroiditis quality of life selenium trial (CATALYST)
PRO endpoints: 1°
‘The primary outcome is thyroid-related quality of life during 12 months’ intervention, as measured by a composite score from the ThyPRO questionnaire. Sample size estimation is based on this outcome.
The trial should be sufficiently powered to identify a difference between the intervention and the control group of 4 points on the 0–100 ThyPRO composite scale, corresponding to a small to moderate effect. In previously obtained data, the SD of ThyPRO-scores (sigma level) was 20 points. With a correlation between observations on the same participant of 0.50, and a power of 80% and a type I error probability (two-sided α level) of 0.05, a sample size of 236 experimental participants and 236 control participants is required. The sample size estimate is based on a design with five repeated measurements having a compound symmetry covariance structure’.47
Trial name: cosmesis and body image after single-port laparoscopic or conventional laparoscopic cholecystectomy: a multicentre double-blinded randomised controlled trial (SPOCC-trial)
PRO endpoints: 1°, 2°
‘The primary endpoint of the study concerns patient’s satisfaction with cosmesis and body image 12 weeks after surgery. This endpoint is assessed using a validated cosmesis and body image score (CBIS) that was previously used in surgery for Crohn’s disease and in donor nephrectomy. This score is calculated on an 8-item multiple choice type questionnaire ranging between 8 and 48 points.
A clinically relevant improvement of the CBIS is defined as an improvement of 20% of the cosmesis score (8 points). Given the reported SD of the CBIS between 4 and 6 and using (alpha=0.05 and beta=0.90), two groups of 49 patients are needed. This is based on a two-sided significance level (alpha) of 0.05 and a power of 0.90. Estimating a 10% dropout rate, which is common in randomised controlled trials, 55 patients will be randomised per arm.’48
Trial group: TROG
Trial name: a randomised phase III study of radiation doses and fractionation schedules for ductal carcinoma in situ of the breast (BIG 3-07/TROG 07.01)
PRO endpoints: 2°
The sample size for this trial, based on the primary endpoint (time to local recurrence of invasive or intraductal breast cancer in the ipsilateral breast), was 1600 patients. The sample size for the PRO substudy was determined a priori, and was less than that required for the primary endpoint, as explained in the protocol excerpt below. Therefore, patients recruited after the PRO-specific target sample size was achieved did not complete PRO questionnaires, saving trial resources in data collection and management.
‘Sample size determination: for the quality of life study aiming to detect a difference between the tumour bed boost and no boost groups of 0.2 SD of a continuous scale such as fatigue or physical symptoms, with 80% power at a two-sided alpha level of 5%, the required sample size is 790 patients. To allow for attrition at a rate of 5% per year, 1020 patients are required to participate in the quality of life study.’49
Explanation
As with any primary endpoint, including those that focus on PRO, the criteria and methods for estimating the necessary sample size should be specified, with adjustments for expected discontinuation from the clinical study.46 Ideally, the criteria for clinical significance (eg, minimal important difference, clinically meaningful within-patient change threshold, responder definition) should be specified when known.50 51 It is important to note that the FDA is more interested in what constitutes a meaningful within-patient change in score from the patient perspective.16
In cases where the PRO is specified as a key secondary endpoint, the statistical power based on the estimated sample size for the primary endpoint, should be determined. If overpowered, specifying a smaller PRO-specific sample size will save trial resources, as illustrated in the BIG 3–07/TROG 07.01 example. When sufficient power may be achieved by collecting PROs from a representative subset of participants, the sampling strategy should be clearly described.
Only 50.7% of NIHR Health Technology Assessment clinical trial protocols address sample size and statistical power for PRO specified as secondary endpoints.3 If the clinical trial is international in scope, the sampling across countries may be influenced by availability of language translations.52 In addition, the variability of measurement between countries may inflate type 2 error (reduces power).
Methods: data collection, management and analysis
SPIRIT-18a(i)-PRO Extension
Justify the PRO instrument to be used and describe domains, number of items, recall period, instrument scaling and scoring (eg, range and direction of scores indicating a good or poor outcome). Evidence of PRO instrument measurement properties, interpretation guidelines and patient acceptability and burden should be provided or cited if available, ideally in the population of interest. State whether the measure will be used in accordance with any user manual and specify and justify deviations if planned.
Examples
Trial name: impact of a multimodal support intervention after a ‘mild’ stroke (YOU CALL-WE CALL)
PRO endpoints: 1°, 2°
‘A number of quality of life tools were reviewed (eg, SF-36, Stroke Impact Scale, Quality of Life Index (QLI)) and the tool chosen was a compromise between psychometric properties and adequacy of content for mild stroke. The 32-item questionnaire QLI (Reference) which was developed from Ferran’s conceptual model of quality of life and which has been used with a stroke clientele (Reference) was chosen as the primary outcome. Each item of the QLI as relating to four life domains (health and functioning, socioeconomic, psychological/spiritual and family), is evaluated in terms of satisfaction and importance on a 6-point scale. Scores for each domain and a global score are expressed from 0 to 30, with a higher score indicating a better quality of life. These four life domains relate well with the main issues covered through the WE CALL intervention. It has shown to have adequate psychometric properties (concurrent validity, test–retest reliability and high internal consistency: a=0.90) (Reference) and thus should be responsive to therapy-induced change (Reference). A 1-point difference was observed in the first 6 months poststroke descriptive follow-up (n=63) for an effect size of 0.33 (Reference). A 2-point difference is considered a clinically meaningful change leading to a moderate effect size of 0.66.’53
Trial name: a randomised phase II/III multicentre clinical trial of definitive chemoradiation, with or without cetuximab, in carcinoma of the oesophagus (SCOPE 1: Study of Chemoradiotherapy in Oesophageal Cancer Plus or Minus Erbitux)
PRO endpoints: 2°
HRQL instruments
‘Generic domains of HRQL will be assessed with the EORTC core Quality of Life Questionnaire, the EORTC QLQ-C30 (Reference). This instrument has been well validated in many international clinical trials in oncology including oesophageal adenocarcinoma and squamous cell cancer. Disease-specific and Chemoradiotherapy (CRT)-associated symptoms and side effects will be assessed with the oesophageal cancer-specific module, the EORTC QLQ-OES18 (Reference). This has been validated and tested in patients receiving definitive CRT. The module includes scales assessing dysphagia, eating restrictions, reflux, dry mouth and problems with saliva and deglutition. The Dermatology Life Quality Index (DLQI) will also be administered (Reference). This is a well-validated, easy-to-use index which assesses the impact of dermatological conditions on patients’ HRQL (Reference). It has been included to accurately assess the impact of the acneiform eruption commonly seen with cetuximab.’54
Explanation
The justification for the selection of PRO instrument(s) is required in the trial protocol. This will help trial personnel and participants understand why specific measures are being used and how they directly address the trial objectives and stakeholder needs.11 For example, regulatory agencies often focus on physical symptoms and functioning to inform licensing and labelling claims, whereas patients and health-policy makers may be more interested in broader aspects of HRQL, such as engaging in social activities and emotional well-being.55 56 For regulatory trials, it is prudent to seek regulatory advice at an early stage of trial development regarding the acceptability of the instrument and the approach to PRO assessment. Stakeholder-relevant PROs can be identified through patient involvement, qualitative research or core outcome sets,56 57 which alongside clinical outcomes, often include outcomes such as symptom burden, functioning, and disease control, which can be measured using PRO instruments.
Appropriately developed and evaluated PRO instruments can provide more sensitive and specific measurements of the effects of medical intervention, thereby increasing the efficiency of clinical trials that attempt to measure the meaningful treatment benefits of those therapies.58–60 Irrespective of whether the trial is conducted for regulatory purposes, FDA guidance and ISOQOL guidance provide a useful conceptual framework to assist in the selection of measures.16 61 Identifying and selecting valid, reliable tools that are acceptable to patients from the target population may prove challenging. The Consensus Based Standards for the Selection of Health Measurement Instruments (COMET) initiative and the Evaluating the Measurement of Patient Reported Outcomes programme provide useful guidance to support the review of measurement properties.56 62 63
Ideally, the PRO instrument(s) will have been validated in the target population and this evidence cited. This will help reviewers understand if claims being supported by the PRO instrument can be substantiated by the evidence for using that instrument in that, or a related, population. Further details on the domains, number of items, recall period, instrument scaling and scoring (eg, range and direction of scores indicating a good or poor outcome) should be provided. This will assist trial personnel in the collection and analysis of the PRO data. Questionnaires should be used in accordance with user manuals to promote good data quality and ensure standardised scoring. Deviations from user manuals or different ways of capturing PRO data may invalidate the measure; therefore, any deviations should be declared and transparently reported.19
If in the trial there are plans to use a questionnaire which has not been validated in the trial’s target population, or if a new instrument is being developed alongside the trial, it is important to explain this in the protocol. Including an outline of any plans for the evaluation of its measurement properties using the trial data, if this will be undertaken, and if not why. This should be in accordance with established current guidelines for PRO validation.64
Although all the reviewed NIHR Health Technology Assessment clinical trial protocols identified the PRO instrument to be used in the trial, few justified their use in relation to the study hypotheses, PRO instrument measurement properties or expected participant burden (41.3%, 37.3% and 14.7%, respectively).3
Patient partners involved in the design of the study can assist with the selection of PRO instruments and provide feedback on the likely acceptability of the questions, and participant burden (eg, time taken for completion, cognitive burden, emotional burden, repetition across questionnaires).65 The number of PRO instruments/questions to be assessed in a trial requires careful justification. Minimising participant burden has been identified as a strategy to reduce risk of missing PRO data, improve recruitment and retention.30
SPIRIT-18a(ii)-PRO Extension
Includes a data collection plan outlining the permitted mode(s) of administration (eg, paper, telephone, electronic, other) and setting (eg, clinic, home, other).
Examples
Trial name: early surgery versus optimal current step-up practice for chronic pancreatitis: a multicentre randomised controlled trial
PRO endpoints: 1°, 2°
‘The Izbicki pain score will be assessed every 2 weeks during a follow-up period of 18 months. For this end, the Izbicki pain score will be assessed via a web questionnaire. Patients who do not have an email will be given a folder with Izbiki pain score forms and return envelops. Patients will be contacted by telephone every 2 weeks and reminded to fill in the questionnaire and send it to the trial coordinators. The Izbicki pain score is a one page questionnaire, easily completed in <3 min. The folder with the Izbicki score forms will be re-filled at every outpatient clinic visit (scheduled every 6 months).’66
Trial name: a randomised multistage phase II/III study of sunitinib comparing temporary cessation with allowing continuation, at the time of maximal radiological response, in the first-line treatment of locally advanced/metastatic renal cancer (STAR)
PRO endpoints: 1°, 2°
Quality of life questionnaires during the first 6 months will be administered in clinic in order to support participant use before postal questionnaires are instituted after 6 months for the EQ-5DTM/EQ-VAS (Functional Assessment of Cancer Therapy - General (FACT-G) and Functional Assessment of Cancer Therapy-Kidney Symptom Index (FSKI) will continued to be collected at clinic visits). Clinic staff should remind participants of the importance of the quality of life assessments at each clinic visit.
Due to the importance of HRQL data in this trial, measures will be taken to ensure maximum compliance of questionnaire completion. For the 2 weekly questionnaires which participants complete at home from the 24-week timepoint, where the participant consents to this, reminders for completion are sent by email or text message to the participant by the research team at the Clinical Trials Research Unit (CTRU): this is an optional part of the STAR Informed Consent Form. Where a HRQL questionnaire would be completed at a hospital clinic visit, but the local research team forget to give this to the participant, of the participant no longer attends clinic visits at hospital during their follow-up period, a questionnaire for the local research team will send this out by post to the participant’s home after checking the participant’s status and establishing it is appropriate to do so.67 68
Explanation
Standardisation of all aspects of PRO administration is vital to PRO data quality. It is therefore critical that research personnel and trial participants understand how, when and where PRO data will be collected in the study.10 The study protocol should specify the permitted mode(s), method(s) and setting(s) of PRO data collection, including the permitted ‘back-up’ options and preplanned reminders. For example, when PRO assessment is conducted in clinic via a tablet computer, paper forms could be permitted (and available) as a back-up option for instances when the tablet is not available or functioning properly. Offering alternative modes of completion may help improve response rates.30 Of note, the FDA has previously recommended that there is a back-up plan for electronic PRO data collection (eg, web-based, phone-based or paper-based) implemented in case of malfunctions with electronic devices.69
Electronic PRO assessment is increasingly available in trials, but traditional paper-based methods may still be useful or required in some situations. It is therefore important to know whether there are systematic differences induced by mode of administration. A recent meta-analysis that included 31 studies that randomised participants to different data collection modes found no evidence of bias associated with paper versus electronic administration.70 These results support the use of multiple modes of administration within a research study, which may be a useful strategy for reducing missing PRO data. If evidence of equivalence between different modes of administration is available for the specific PRO questionnaires in a trial, it should be considered in determining the PRO administration plan. If electronic administration has not been attempted before for the trial PRO questionnaires, and only minor modifications to layout/presentation are needed with respect to the paper-based versions, it is advisable to pilot-test usability and conduct cognitive debriefing to assess equivalence.70 71 The International Society for Pharmacoeconomics and Outcomes Research provide useful guidance on key considerations for PRO data collection including multiple modes.71 72
The setting for PRO data collection, for example, in clinic or at home (or clinic at baseline, with follow-up at home), should be described and standardised across trial intervention groups and sites. Differential use of settings and modes of administration by treatment arm should be avoided as these may lead to different response rates and potentially biased results.11
The protocol should also specify the types of assistance trial staff can provide patients for completing the PRO assessment. Respondents should be encouraged to self-complete as far as possible. Some respondents may require some assistance, however, the greater the degree of assistance, the greater the potential to influence a respondent’s responses. Assistance should therefore be limited, provided only by a trained member of the research team, or a trained third party, the permissible types of assistance should be clearly specified in the protocol and reviewed in staff training. Allowable assistance might include instructions on how patients can input their answer on the tablet, clarifying the response options, reading questions to the participant or recording the participants’ answer on the form/tablet. This level of assistance facilitates self-administration of the PRO instrument. Completion of a PRO instrument with an interpreter, caregiver or family member should be avoided as these individuals have not been trained, and may influence the individual’s responses, either directly by expressing opinions that influence the participant to alter their answers, or indirectly, for example, if the respondent seeks to avoid embarrassment or to provide a more acceptable answer (social desirability bias).
Furthermore, use of a human language interpreter should be avoided. When planning a study, common languages spoken by patients attending the recruiting centres should be considered so that validated language translations of chosen PRO instruments can be obtained (see SPIRIT-18a(iii)-PRO Extension).
Interviewer administration of PRO instruments should be avoided, but where necessary, should be clearly justified in the protocol. Interviewers should read questions verbatim, ideally using a PRO instrument that has been validated in that mode. Similarly, proxy or observer completion requires a proxy-validated/observer-reported version of the PRO instrument (see SPIRIT-18a(iv)-PRO Extension).
SPIRIT-18a(iii)-PRO Extension
Specify whether more than one language version will be used and state whether translated versions have been developed using currently recommended methods.
Example
Trial name: A Phase III, randomised, open label trial of lenalidomide/dexamethasone with or without elotuzumab in relapsed or refractory multiple myeloma (ELOQUENT – 2)
PRO endpoints: 2°, exploratory
Outcomes research assessments
HRQL assessments
‘To assess the impact of treatment, subject’s quality of life will be measured using three validated HRQL instruments: the European Organization for Research and Treatment of Cancer Quality of life Questionnaire-Core (EORTC QLQ-C30), the myeloma-specific module (QLQ-MY20) and the Brief Pain Inventory-Short Form (BPI-SF).
Non-English-speaking subjects will complete the questionnaire using validated language transitions developed and recommended for each instrument. The BPI-SF has demonstrated both reliability and validity across cultures and languages, and has been used to study the effectiveness of pain treatment.(Reference) A score of 6 on a scale of 0–10 on any single item is generally considered to be clinically significant.(Reference) Pretesting was carried out in the UK, Norway, Sweden, Denmark and Germany. Field testing of the module has been conducted in a range of phase III trials.(Reference) The module has been validated in a large number of languages (see www.eortc.be/home/qol).’73
Explanation
Trials involving participants with different language requirements require measures that have been translated and culturally adapted using appropriate methodology.10 12 74 75 Providing culture-appropriate and language-appropriate PRO instruments for use in the trial can lead to a reduction in missing data, ability to recruit people from ethnic minority groups, lower attrition rates and improved generalisability of trial results.76 If the countries/languages are not known at the time of protocol writing then more general protocol content may be appropriate:
multiple language validated versions are available [provide references where these can be found] and the correct language for this patient should be used.
At present, the extent to which this is happening is not clear. A review of protocols and/or subsequent publications from cancer clinical trials with a PRO endpoint, registered on the NIHR portfolio examined reporting of ethnically diverse recruitment and the use of culturally and linguistically validated PRO instruments. The review found a lack of transparency around the use of culturally and linguistically appropriate PRO instruments. Of the 88 studies reviewed, only 14 (17%) reported any type of data on ethnic diversity. Although eight studies were multicentre, multinational cancer clinical trials, none identified if translated versions of PRO instruments were being used.77
There are clear guidelines for translating PRO instruments,74 78 and plans to use translated versions, should be specified in the protocol, citing references when available.10 Specification of use of translated versions in the protocol will help reporting in accordance with CONSORT-PRO.19 74 75 It must not be assumed that linguistic translation equates to cross-cultural adaptation (preparing the instrument for use in another setting). A number of studies79 80 have recommended that cross-cultural equivalence is also an important consideration.74 75 81
Using different language versions of PRO instruments to collect data in a trial ideally requires evidence to support the psychometric equivalence of data being reported, especially if data are going to be pooled for clinical trial evaluation.52 82 83 Where such evidence is unavailable, prespecification in the statistical analysis plan (SAP) of exploratory analyses to assess whether there are differences between PROs by language group may be appropriate. The language in which each patient complete the questionnaire should be recorded in the database to inform such analyses.52
SPIRIT-18a(iv)-PRO Extension
When the trial context requires someone other than a trial participant to answer on his or her behalf (a proxy-reported outcome), state and justify the use of a proxy respondent. Provide or cite evidence of the validity of proxy assessment if available.
Examples
Trial name: cognitive rehabilitation in paediatric acquired brain injury—a randomised controlled trial (CORE-pABI)
PRO endpoints: 1°, 2°
‘[Paediatric Acquired Brain Injury] constitutes a major disruption to child development and may affect cognitive, behavioural, emotional, social as well as academic function.
The primary outcome measure is the BRIEF, parent report (Reference). BRIEF is an 86-item standardised questionnaire that captures parents perceptions of a child’s executive function in his or her everyday environment. Each item’s frequency of occurrence is rated on a 3-point Likert scale from 1 (never) to 3 (often). It has demonstrated good reliability, with high test–retest reliability (r=0.88 for teachers,82 for parents), internal consistency (Cronbach’s α=0.80–0.98) and moderate correlations have been detected between teacher and parent ratings (r=0.32–0.34). The questionnaire has been applied to several clinical groups in Norway.’84
Trial name: evaluating the effectiveness and cost effectiveness of dementia care mapping (DCM) to enable person-centred care for people with dementia and their carers: a cluster randomised controlled trial in care homes (DCM EPIC study 1.0)
PRO endpoints: 2°
‘Relative/friend criteria
To be eligible to provide proxy data about a resident, relatives/friends must: have visited the resident on a regular basis over the past month (ie, at least once per week). Be willing to provide data at a time convenient to them. Have sufficient proficiency in English to contribute to the data collection required for the research.’85
Explanation
In some contexts, such as trials involving young children or cognitively impaired participants or participants who are unable to reliably self-report for other reasons, it may be necessary for a proxy—someone other than a trial participant, to report the participant’s outcomes on their behalf as though they are the patient.10 86
Proxy reports should be used only when necessary. The European Medicines Agency states that ‘in general proxy reporting should be avoided, unless the use of such “proxy raters” may be the only effective means of obtaining information that might otherwise be lost.’16 43 The US FDA also discourages the use of proxy-reported outcomes to inform labelling claims, recommending observer reports for observable phenomenon only (eg, vomiting, but not nausea) instead.
In contexts such as cancer, dementia or palliative care, it is reasonable to anticipate the need for proxy response throughout all or some of the trial. Previous studies have shown varying levels of agreement between participant and proxy ratings, dependent on the variable being measured, the quality, duration and stability of the relationship between proxy and participant.87 88
A trial protocol should indicate clearly who is eligible to provide the proxy report, with explicit administration guidelines for completion of proxy measures including how the report is to be captured, whether that same individual must be the ‘consistent rater’ across all timepoints of assessment (this is preferable, for consistency), or whether varying proxy reports will be permissible. This information should also be provided for observer-reported outcomes.
Just as the measurement properties of the PRO instrument should be specified, so should the properties of measures to be used by proxy reporters.
Given known issues with patient and proxy reporter discordance,88–91 while patient-participants are still able to self-complete, collecting both participant and proxy-reported data enables quantification of the size and direction of any bias, that may later be adjusted for, if needed. Further data may be gathered about the proxy (eg, age, relationship to the patient, gender, proxy literacy, relationship and exposure to the patient92) as these variables may guide interpretation of results and any subgroup/sensitivity analyses. Whether proxy-reported data will be analysed separately or pooled with participant-reported data should also be detailed. Any such plans should be specified in the protocol and SAP. This information should also be provided for observer-reported outcomes.
SPIRIT-18b(i)-PRO Extension
Specify PRO data collection and management strategies for minimising avoidable missing data.
Examples
Trial group: National Cancer Institute, Naples
Trial name: phase III randomised multicentre trial of carboplatin+liposomal doxorubicin versus carboplatin+paclitaxel in patients with ovarian cancer (Multicentre Italian Trials in Ovarian Cancer-2 (MITO-2))
PRO endpoints: 2°
‘Operating procedure:
It is fundamental that the researchers take great care when collecting the questionnaires, in order to allow good compliance by the patients participating in the protocol.
The quality of life form must be filled in by the patient herself.
The quality of life form must be filled in before the clinical examination, and thus before the discussion with the examining doctor which may provide favourable or unfavourable information about the disease’s status.
When supplying the form to the patient, it is important to explain how to fill it in without going into details about the contents of the questions.
After the form has been returned, check that the patient has answered all the questions and ask her to reply to any questions she has skipped.
The quality of life questionnaires must be filled in using a black or blue pen.’93
Trial group: National Cancer Institute of Canada Clinical Trials Group (NCIC CTG)
Trial name: a double-blind randomisation to letrozole or placebo for women previously diagnosed with primary breast cancer completing 5 years of adjuvant aromatase inhibitor either as initial therapy or after tamoxifen (including those in the MA.17 Study) (NCIC CTG: MA.17R)
PRO endpoints: 2°
Quality of life
‘Mandatory for NCIC CTG centres and optional for centers within other cooperative groups:
Patient is able (ie, sufficiently fluent) and willing to complete the two quality of life questionnaires in either English or French. The baseline assessment must have been completed prior to randomisation. Inability (illiteracy in English or French, loss of sight or other equivalent reason) to complete questionnaires will not make the patient ineligible for the study. However, ability but unwillingness to complete the questionnaires will make the patient ineligible.’94
Explanation
Missing PRO data are a particular problem because data cannot be obtained retrospectively or from medical records. Missing PRO data may arise from different sources95 and, broadly speaking, missing data can be attributed to causes that are unavoidable or avoidable. Unavoidable reasons may include if a participant has died or become too unwell to self-complete PRO instruments. Avoidable reasons may include some type of human error that could have been prevented. Examples of avoidable missing data include: staff failing to hand out a scheduled questionnaire; a participant not realising the questionnaire is double-sided so missing half of the questions; an electronic PRO device not being charged; the internet or server being down; when the PRO assessment is overly burdensome on the patient (eg, due to the burden of multiple questionnaires at one time or repetitive scales) so the patient decides not to complete it.
Although ‘unavoidable’ types of missing data are more challenging for interpretation because the missing data may be related to the measured outcome and it is impossible to accurately calculate the extent of any associated bias,96 97 avoidable types of missing data are also problematic.30 Avoidable missing PRO data compromise the interpretability, accuracy and value of PRO findings because study power is reduced, which increases the risk of type 2 errors,96 and because any assumptions made during the analysis about missing PRO values are not verifiable.98
There are a range of design, implementation and reporting strategies to help minimise and address missing PRO data,30 most of which can be addressed in the trial protocol. Specific recommendations related to data collection and management include: refraining from administering an excessive number of questionnaires to participants (researchers should refrain from collecting more data than they really need), using standardised and documented PRO administration procedures, engaging and educating participants in the trial by providing updates or incentives, maintaining participant records, employing active quality assurance measures (such as real-time compliance/completion monitoring, sending reminders for upcoming or missed assessments, checking completed questionnaires for missing items while the participant is still present at clinic), appointing a dedicated staff member responsible for PRO assessment at each centre, training staff about the importance of PROs as well as procedures for assessment, offering an alternative mode of administration if the participant is not able to complete the questionnaire via the primary mode (eg, completing the questionnaire over the phone if the hardcopy cannot be completed at the clinic within the acceptable time-window) and recording reasons for missed assessments using standardised forms.30 From a regulatory perspective, the FDA encourages maintaining consistency in assessment methods, however, this should be balanced with reduction of missing data. If different modes are used, they should be justified and presented in the study documentation.99 If different modes are used, ‘FDA will review the comparability of data obtained when using multiple data collection methods or administration modes within a single clinical trial to determine whether the treatment effect varies by method or mode.’69 Additional strategies are described in the full review.30
The MITO-2 and MA.17R examples above illustrate some of these strategies. MA.17R comes from the Canadian Cancer Trials Group (CCTG), a multicentre cooperative oncology group that conducts clinical trials in cancer therapy, supportive care and prevention. The CCTG requires completion of the PRO questionnaire(s) as a prerandomisation eligibility requirement (as per SPIRIT-10-PRO Extension). This flags the importance of PRO data to investigators and clinical research associates, indicating PROs are as important as other inclusion clinical criteria. It also helps to maximise compliance.
Our prior work suggests that few trials actively specify such procedures in their protocols; 46.7% HTA protocols3 and 38.5% of international ovarian cancer trials4 included strategies to minimise avoidable missing data.
SPIRIT-18b(ii)-PRO Elaboration
Describe the process of PRO assessment for participants who discontinue or deviate from the assigned intervention protocol.
Examples
Trial name: a multicentre, open-label, randomised, two-arm phase III trial on the effect on PFS of bevacizumab plus chemotherapy versus chemotherapy alone in patients with platinum-resistant, epithelial ovarian, fallopian tube or primary peritoneal cancer (AURELIA)
PRO endpoints: 2°
AURELIA was a multicentre, open-label, randomised, two-arm phase III trial of bevacizumab plus chemotherapy versus chemotherapy alone in patients with platinum-resistant, epithelial ovarian, fallopian tube or primary peritoneal cancer (Reference). The primary endpoint was PFS; quality of life was a secondary endpoint. The protocol stated that:
‘On clear evidence of disease progression (PD) or toxicity, study therapy should be discontinued permanently.’
The protocol also specified postprogression treatment options: women who had been on the chemotherapy alone arm would have the option of bevacizumab alone or standard of care, while those who had been on the chemotherapy plus bevacizumab arm would receive standard of care treatment. The protocol stated that:
‘In case the patient decides to prematurely discontinue study treatment (“refuses treatment”), she should be asked if she can still be contacted for further information’ and that ‘after PD, patients will be followed for survival only.’
However, it lacked an explicit statement about PRO assessment postprogression, which may explain some inconsistency among sites, with some sites collecting PRO data postprogression, and other not.
As stated in the AURELIA PRO paper (reference, p1310), ‘only questionnaires completed until PD were included in the main analyses. Questionnaires completed after PD were excluded based on the medical assumption that these patients were unlikely to be benefiting from their study treatment, may have been receiving another treatment and were therefore not relevant to the intended comparison of chemotherapy alone versus bevacizumab plus chemotherapy. However, post hoc sensitivity analyses were performed to determine the impact of questionnaires completed after PD.’ The latter analyses were consistent with the main analyses, but could perhaps have been avoided, and patients saved unnecessary HRQL assessment burden, if the protocol has contained a clear statement that HRQL assessments should cease at disease progression.100
Trial group: Australasian Leukaemia & Lymphoma Group (ALLG)
Trial name: BLAM-A phase IIb study of blinatumomab+cytarabine (AraC) and methotrexate in adult B-precursor acute lymphoblastic leukaemia (ALLG ALL8)
PRO endpoints: 2°
ALLG ALL8 is a BLAM-A phase IIb study of blinatumomab+cytarabine (AraC) and methotrexate in adult B-precursor acute lymphoblastic leukaemia (B-ALL). It is a single-arm study which aims to demonstrate preliminary evidence of the benefit of frontline blinatumomab in combination with cytarabine and methotrexate in adult B-ALL, and to demonstrate the ability of this combination to attain deep (MRDnegative) remissions and hence to reduce the need for allogeneic stem cell transplantation. The ALLG ALL8 protocol specifies that the decision for allogeneic stem cell transplantation is left up to the investigator, and that patients who proceed to allogeneic stem cell transplant discontinue the assigned intervention protocol. Being recommended for allogeneic stem cell transplant is therefore a withdrawal criteria. However, patients do not have to withdraw—it is optional, the patient’s choice. The trial team is in fact interested in the experience of patients who proceed to allogeneic stem cell transplant. So the protocol states:
Subjects who proceed to allogeneic stem cell transplant who have not withdrawn consent to the study should have FACT-Leu (HRQL questionnaire) assessments performed 6-monthly.
This provides the trial team with the opportunity to document the experience of patients who proceed to allogeneic stem cell transplant in terms of the range of common symptoms and aspects of well-being assessed by FACT-Leu.101
Explanation
A clear plan for collection of PROs for trial participants who withdraw early from a study or who discontinue the intervention helps minimise bias,102 ensures that staff collect all required PRO data in a standardised and timely way, and may assist ethical appraisal of the study.10 102
Often, participants can provide valuable PRO data even after stopping the assigned intervention protocol, whether due to personal choice and/or clinical recommendation, as illustrated in the one-arm phase II trial ALLG ALL8 trial. However, this does not hold in all contexts (as illustrated in the AURELIA trial), a randomised phase III trial, in which participants whose cancer had progressed on their assigned treatments, were then often switched to alternative treatment.
Providing a clear description of the process of PRO assessment for participants who discontinue or deviate from the assigned intervention protocol and how the data will be used, enables all staff to follow a standardised procedure to collect the required PRO data in timely way and to avoid collecting data that will not contribute to analysis.
Correspondingly, the SAP should be clear on how such data will be handled. In the case where postdiscontinuation/deviation PRO data are useful, the SAP should state the study objective that these data will address and how they will be analysed.
Participants should also be aware of this process, so a simple and clear description of whether or not they will be asked to continue to complete PRO questionnaires after stopping or changing the treatment they were initially allocated to should be included in the participant information sheet (PIS).
SPIRIT-20a-PRO Elaboration
State PRO analysis methods, including any plans for addressing multiplicity/type I (α) error.
Example
Trial name: a phase III, randomised double-blind, placebo-controlled study of the efficacy and safety of two doses of tofacitinib (Cp-690,550) in subjects with active psoriatic arthritis and an inadequate response to at least one tumour necrosis factor inhibitor (OPAL BEYOND)
PRO endpoints: 1°, 2°, exploratory
Analysis of primary endpoint
‘There are two primary endpoints in A3921125 and two doses of tofacitinib each of which will be compared with placebo for each endpoint. In order to control for type I error, a stepwise testing procedure will be used. This implies that a given endpoint for a given dose can only achieve significance if the prior endpoint is significant. The order of the fixed sequence for testing against placebo is as follows: tofacitinib 10 mg ACR20 response rate at 3 months, tofacitinib 5 mg ACR20 response rate at 3 months, tofacitinib 10 mg Health Assessment Questionnaire-Disability Index (HAQ-DI) at 3 months, tofacitinib 5 mg HAQ-DI at 3 months. This gate-keeping or step-down approach strongly protects the type I error rate at the 0.05 (two-sided) level.
ACR20 response
For all comparative analyses, the normal approximation for the difference in binomial proportions will be used to test the superiority of each dose of tofacitinib to placebo and to generate CIs for the differences. The primary analysis will be ACR20 response rate at month 3. The ACR20 response rate will also be analysed at other timepoints as a secondary analysis.
Missing values due to a subject dropping from the study for any reason (eg, lack of efficacy or adverse event) will be handled by setting the ACR20 value to non-responsive.
HAQ-DI
The HAQ-DI score will be expressed as a change from baseline. The primary timepoint will be at month 3. The analysis will be done using a repeated measure model that includes fixed effects of treatment, visit (week 2, months 1, 2 and 3), treatment by visit interaction, geographic location and baseline value. The model will use and fit an unstructured variance-covariance matrix. Full details will be listed in the analysis plan.
Additional analyses of the HAQ-DI will include a responder analysis at month 3 where subjects with a change of 0.3 will be considered responders and subjects who dropped from the study will be considered non-responsive. Another responder analysis will be conducted using a change of 0.35 at the cutpoint for response (Reference). The normal approximation for the difference in binomial proportions will be used for these responder analyses.
Analysis of secondary and other endpoints
Key secondary efficacy variables are as follows: PASI75, enthesitis score, dactylitis severity score, physical function domain of SF-36 and FACIT-F at month 3. In order to strongly protect the study-wise type I error rate with respect to these key secondary endpoints and the primary endpoints, these endpoints will be tested only if all endpoints/doses for the primary endpoints are statistically significant. The order of testing is as listed above; for each endpoint, tofacitinib 10 mg will be tested versus placebo first, followed by tofacitinib 5 mg. Testing stops at the first instance in which statistical significance is not achieved.
Methods for analysing all other endpoints will be enumerated in the statistical analysis plan. Briefly, binary variables (eg, remission rates) will follow the analyses described above for binary variables (eg, ACR20) and continuous endpoints will follow the same type of analyses described above for continuous endpoints (eg, HAQ-DI). Descriptive statistics may also be calculated and displayed’.103
Explanation
Statistical analysis of multiple domains10 104 and timepoints implies multiple hypothesis testing, which inflates the probability of false-positive results (type I error).46 This can be contained by prespecifying the key PRO domain(s) or overall score of interest and the principal timepoint(s) which cross-reference to SPIRIT-7-PRO Extension and SPIRIT-12-PRO Extension.
Any plans to address multiplicity, such as stepwise or sequential analyses, whereby multiple endpoints are tested in a defined sequence that contains the overall type I error to the desired level, or conventional non-hierarchical methods (eg, Bonferroni correction), should be specified a priori.16 There are many strategies and/or choices of methods that may be appropriate.105 Family-wise type I error should be considered for all of the applicable endpoints of the trials together and not for the PRO endpoints separately. Some clinical trials include PROs as exploratory endpoints and no adjustment is made for multiplicity in subscale scores administered at multiple study visits. These analyses may only provide limited information on the tolerability of the intervention.
Protocols should make some reference to key considerations for the analysis of the trial PROs (including any plans for addressing multiple testing), but the detail is often more appropriately included in the SAP, usually developed after the protocol. If no adjustments for type I error are to be made, then this should be clearly stated. However, clinical trial protocols in which PROs are secondary outcomes rarely include any information about PRO statistical analyses, beyond any that are prespecified as primary or key secondary endpoints or when the sponsor is interested in achieving a product label claim. Our review of trial protocols found that fewer than 2% provided information on the statistical analysis plans to address multiplicity for the PRO endpoints.3
SPIRIT-20c-PRO Elaboration
State how missing data will be described and outline the methods for handling missing items or entire assessments (eg, approach to imputation and sensitivity analyses).
Example
Trial group: European Organisation for Research and Treatment of Cancer-Gynaecological Cancer Group, and Gynecologic Cancer Intergroup (EORTC-GCG and GCIG)
Trial name: a randomised, multicentre, phase III study of erlotinib versus observation in patients with no evidence of disease progression after first line, platinum-based chemotherapy for high-risk ovarian epithelial, primary peritoneal, or fallopian tube cancer (EORTC 55041)
PRO endpoints: 2°
Compliance
‘Missing data may hamper assessment of HRQL in clinical trials. This may occur because centres do not collect the questionnaires at the appropriate time (unit non-response), and also because patients may not reply to questions within the questionnaires (item non-response). The latter problem occurs <2% on average and should not be a problem. The former problem will be minimised by ensuring that participating centres are properly informed and motivated towards HRQL assessment.
During the study, compliance with completing HRQL questionnaires will be investigated at each timepoint. The compliance of the HRQL assessments will also be reviewed twice a year and will be a part of the descriptive report by the Data Center for the Group’s plenary sessions and, if possible, be presented by the EORTC Quality of Life Group’s appointed liaison person.
The compliance rate between the two arms will be compared at each timepoint using a χ2 test. In order to adjust for the multiplicity of the tests, a Bonferroni adjustment will be made by which each test will be performed at the 0.01 significance level. Should follow-up compliance levels drop below 60% at subsequent biannual compliance reviews, then the Protocol Writing Committee would review this to either improve compliance or consider terminating the HRQL assessment in the trial.
Missing data
When performing HRQL analyses complications may arise due to large quantities of missing data. This issue has a bearing on whether a valid comparison of the treatment arms is being made.
In HRQL research there are two main types of missing data: (1) item non-response, (2) unit non-response (the whole questionnaire is missing for a patient). As item non-response occurs <2% on average in the QLQ-C30, it is not such a major problem and thus the methods described in the EORTC QLQ-C30 scoring manual for handling item non-response will be used. For missing questionnaires, it is necessary to identify both the extent of missing questionnaires and the main process of missing data. Three different types of missing data processes may exist: missing completely at random (MCAR), missing at random (MAR) and missing not at random (MNAR, informative dropout mechanism). These have distinct consequences for data analysis (Reference).
If the missing data process is considered to be non-ignorable (MNAR), then quality of life will be compared between groups using longitudinal data modelling techniques (ie, Proc mixed in SAS with either selection models or pattern-mixture models) in combination with a logistic regression for the dropout process. If the missing data mechanism can be considered ignorable (MAR), then standard longitudinal data analysis will be used (proc mixed in SAS). If the data are MCAR, then complete case analysis can be used without biasing the results.’106
Explanation
Most clinical trials with PROs will have some missing PRO data,98 yet a review of protocols found that less than half outlined statistical methods to deal with missing PRO data.3
There are two types of missing PRO data: (1) missing items, when the participant completes some but not all of the questions within a PRO instrument and (2) missing assessments, when the participant does not complete a scheduled assessment at all (ie, there are no PRO data available for analysis from the participant at that assessment timepoint). The latter type is more serious, as it potentially affects the choice of analysis method and the interpretation and generalisability of the results. The trial protocol should explain how both types of missing data will be handled in the analysis.
Handling missing items: many PRO scoring manuals provide guidance for handling missing items. A common approach is to impute the mean score of the completed items, if less than one-half of items comprising the scale are missing.107 108 This approach is possible for multi-item scales but it is not possible to impute scores for single item scales. Always consult the scoring manual to determine how to handle missing items, and cite the manual in the protocol. Missing items of PRO instruments that are underpinned by modern psychometrics techniques (Item Response Theory or Rasch Measurement Theory) are naturally handled without requiring imputation.34 109
Handling missed assessments: missing assessments present a major problem for PRO analyses; lead to a loss of power and wider CIs from a lack of precision,110 111 and potentially to biased results. The risk of bias depends on the underlying cause of why data are missing (called the ‘missing data mechanism’), and this in turn should influence the choice of statistical analysis methods.
In order to gain insights into the mechanism of missing PRO data, it is helpful to ascertain and record reasons for missed PRO assessments during trial conduct. The protocol needs to describe how these reasons will be collected in a standardised manner. Typically, a standard form is used (which can be included in the online supplemental appendix along with the PRO questionnaires). Also, the form can be used to collect additional data (referred to as ‘auxiliary data’) related to ‘missingness’ or the PRO,30 which should be specified in the protocol.
The trial protocol should also provide a summary of how missing data will be described and handled in the analysis,10 and state that comprehensive details about the planned analysis will be provided in a subsequent SAP.112 The SISAQOL Consortium have developed a taxonomy of research objectives that can be matched with appropriate statistical methods for PRO analysis, standardised statistical terminology relating to missing data and are determining appropriate ways to manage missing data, currently focused in an oncology setting. A simulation study was done to assess whether it was possible to have a threshold to define substantial missing data.113 Although no agreement was reached for a threshold, the simulation study showed that the effect of missing data rates on PRO findings depends on the type of missing data (ie, informative or non-informative missing data). It was recommended that collecting reasons for missing data is key in assessing the effect of missing data for PRO findings.34 Additionally, SISAQOL is developing a set of macros to describe patterns of missing data, and to evaluate imputation methods for use in sensitivity analysis.34 36 38
Methods: monitoring
SPIRIT-22-PRO Extension
State whether or not PRO data will be monitored during the study to inform the clinical care of individual trial participants and, if so, how this will be managed in a standardised way. Describe how this process will be explained to participants, for example, in the PIS and consent form.
Example
Trial name: the use of an electronic patient-reported outcome measure in the management of patients with advanced chronic kidney disease (RePROM)
PRO endpoints: 2°
Protocol
‘If, when reviewing the completed EQ-5D-5L questionnaire, the research nurse becomes concerned for the well-being of the participant, they should discuss their concerns with the participant directly, working in partnership to determine the best course of action. With the participant’s permission, the research nurse may need to consult with the PI and/or treating clinician to address these concerns. In exceptional circumstances, the research nurse may consult with the PI and/or treating clinician without the permission of the participant if they are concerned for the participant’s safety’.
Patient information sheet
‘If the study research nurse becomes concerned for your well-being during a study review, they will discuss their concerns with you, to determine the best course of action. With your permission, the research nurse may need to consult with a senior member of the study and/or your treating clinician to address these concerns. In exceptional circumstances, the research nurse may need to do this without your prior permission if they are concerned for your safety’.
Consent form
‘I understand that if the study research nurse becomes concerned for my well-being during a study review, they will discuss their concerns with me to determine the best course of action. With my permission, the research nurse may need to consult with a senior member of the study and/or my treating clinician to address these concerns. In exceptional circumstances, the research nurse may need to do this without my prior permission if they are concerned for my safety.’114
Explanation
To protect participant safety, PRO data may be monitored during a study for signs of psychological distress or physical symptoms that may require an immediate response: so-called ‘PRO Alerts’.115 Examples of PRO data that may raise concern include signs of psychological distress, poor physical well-being or high symptom burden presenting as extreme scores on questionnaires. Concerns can also arise when additional information is provided by the participant (eg, through free text report), or in discussions between the participant and research staff.115 The nature of some studies may mean that participants are more at risk than in others. In studies where prior risk assessment deems the probability of PRO alerts being generated by participants to be minimal, PRO data may not be reviewed until the end of the trial for pragmatic reasons, however, concerning PRO data, may still arise during the course of the study.11 116 If monitoring is not planned this should be stated. If monitoring is planned, steps for how PRO alerts will be dealt with should be included in the trial protocol, to provide immediate reassurance to concerned trial staff about how to proceed and promote appropriate clinical management and transparency considering professional obligations to patient care. Any arising interventions should be recorded. Evidence suggests the absence of such information leads to inconsistent handling of concerning data including administration of non-prespecified interventions to aid the trial participant which risks co-intervention bias that is, bias caused by ‘any intervention other than the experimental manoeuver that alters the frequency of a trial’s outcome of interest’.12 116 117 Identifying participants at risk and in need of urgent attention through PRO monitoring is an ethical issue as is acknowledging that information identified in the completion of a PRO too may be shared with the clinical team when the need arises. Information about how the trial staff will respond to concerns, or alternative support mechanisms where monitoring is not taking place, should be provided to participants (in participant information and consent documentation). This information provided in the patient information sheet (PIS) will manage participant expectations and ensure transparent and explicit communication about the intended use of PRO study data in fulfilment of contemporary data protection laws, for example, the European Union General Data Protection Regulation.118
There is no regulatory requirement from the US FDA that PRO data measuring symptomatic side effects be monitored and alerts for these items created. However, it is good practice to remind participants at each PRO assessment whether their data are or is not being monitored in real time. In the case PRO data are not being monitored, participants should be reminded to speak to clinical staff if they are experiencing a specific problem, symptom or side effect.
Box regulatory/HTA perspective
Regulatory agencies, such as the Medicines and Healthcare products Regulatory Agency, European Medicines Agency and FDA are placing more focus on capturing the patient experience when developing drugs.16 43 55 133 However, poorly defined PRO objectives have hindered the utility of PROs in regulatory decisions. Accurate and well-defined PRO methods can provide the patients’ perspective on the impact of a treatment on disease-related symptoms and symptomatic adverse events. Efforts to improve PRO clinical trial standards are welcomed and the SPIRIT-PRO extension provides an additional resource for drug developers to consider in their development programmes. Alongside these guidelines and other regulatory guidance documents, the importance of seeking early scientific advice directly from the regulatory authorities and health technology assessment bodies such as NICE cannot be understated, where advice can be given on the acceptability of a particular approach. Therefore, raising PRO standards is key to the successful integration of PROs in drug development programmes, ensuring that the impact of medicines on a disease can be captured from the patient’s perspectives.
Box patient perspective
What is the question that doctors and nurses ask their patients more than any other? “How are you feeling?” That is why we need PROs in clinical research. They allow patients to answer that question in a systematic and measurable way, which will benefit others and from which we may well benefit ourselves at some point.
For patients, including PROs in health and social care research studies is vital in assessing whether or not our health and/or well-being are improving. Well-designed PRO instruments will help assess our general health and emotional mood, our ability to complete our daily tasks and our self-measured levels of pain and/or fatigue.
PROs are very different from the clinical measures used to assess the effectiveness of new drugs or other treatments, yet for us, it is the PROs that measure ‘tolerability’ and thus the real-life ‘effectiveness’ of the drug or intervention as a medicine or treatment.
The ultimate measure of the performance of any health service must be in whether or not it helps people recover from an acute illness, live well with a chronic condition and face the end of life with dignity—and people’s own reports on their own condition are the only valid way to gauge success. So if a drug or treatment is to be trialled for use in healthcare delivery, it is essential that PROs are included in the success criteria.
It is equally essential therefore that participants on the study understand the importance of completing PRO assessments and to understand how and why the data are collected. This should not be too onerous for researchers to explain to patients. Patients who choose to participate in clinical trials do so because they wish to benefit others and if possible, to benefit themselves. PROs are a means of doing both—provided that the reporting and recording are not too much of a burden. “We must do all that we can to make patient-reported outcome assessment feasible and credible. If we fail in our task we will have left out the heart of all healthcare research: the patient”.141
And when we do complete our PROs on your research study, please let us know what happens to the study, what you know now that you did not know before, and how that will be used to help people. Because that is why we participate in research; it is to help people like us.
Protocol template
The PRO protocol template (online supplemental file 2) aims to support protocol writing for pharmaceutical companies, funders, clinicians and international trials groups by providing PRO content that can be incorporated directly into the relevant sections of existing clinical trial protocols or retained as a dedicated PRO section within the protocol.
Supplementary trial documents
Online supplemental 2 outlines additional items recommended for inclusion in other trial documentation, such as the SAP, PIS and training and guidance documents for staff.10 119 This is not an exhaustive list and further PRO content may be warranted in training materials and patient facing documents dependant on the trial. We recommend input from PRO experts working in conjunction with the clinical team, trials unit or Contract Research Organisation and patient partners involved in the co-design of research with regulatory input as required to optimise the protocol and supplementary resources.
Supplemental material
Discussion
This paper provides a detailed rationale, implementation instructions and real-world examples to assist investigators to develop the PRO-specific components of clinical trial protocols, in accordance with the 16 items of the SPIRIT-PRO Extension. This SPIRIT-PRO Extension E&E paper is recommended for use alongside the original SPIRIT 2013 and SPIRIT-PRO guidelines.1 2 10 The mission of the SPIRIT-PRO Group is to improve the design and standardisation of PRO components of clinical trials and thereby ensure high-quality PRO data to inform patient-centred care. To further facilitate uptake of the SPIRIT-PRO items, we have provided a PRO-specific protocol template covering all the SPIRIT-PRO items. This can be used in two ways: either incorporated item-by-item into relevant sections of existing clinical trial protocols or retained whole as a dedicated PRO section within a trial protocol. The use of a template should support investigators to address all required SPIRIT-PRO checklist items comprehensively and meaningfully, in conjunction with the real-world examples provided for each SPIRIT-PRO item in this manuscript.
The overall aim of the SPIRIT-PRO Extension and E&E is to improve the completeness, quality and transparency of PRO sections of clinical trial protocols, where PROs are a primary or key secondary outcome. We also recommend use of the guidelines to support development of protocols where PROs data are exploratory in nature, including single-arm trials with PRO endpoints. Many of the SPIRIT-PRO items may also provide useful prompts about PRO content for cohort studies and other non-randomised designs. The SPIRIT-PRO guidelines,10 and E&E paper aim to facilitate development of high-quality PRO protocol content, which will ultimately also facilitate the review of protocols by research ethics committees/IRBs, scientific review groups and funders. Improved PRO protocol content has been associated with more complete reporting which will help facilitate the critical appraisal of final trial reports and results3 and use of PRO data to inform patient-centred care. Several SPIRIT-PRO items correspond to items on the CONSORT-PRO checklist.10 19 This is particularly important since reviews of PRO reporting indicate that, where published PRO trial data were available, there was often considerable delay between publication of primary outcomes and the PRO results and standards of reporting were poor.4 5 7 9 120–123 Worryingly, a recent review of cancer trials suggested that 49 568 participants were involved in studies that failed to publish their PRO data and that poor reporting was associated with suboptimal PRO protocol content.9 This finding is consistent with findings from Schandelmaier et al, which demonstrated that 52% of cancer trials specified HRQL outcomes in their protocols, however, only 20% reported any HRQL data in associated publications.120 Non-reporting of PRO findings is widespread,7 9 120–128 meaning patient-centred information may not be available to benefit patients, clinicians and regulators. Non-reporting of these important patient data is unethical and is a waste of limited healthcare research resources.129–131 In the EU Clinical Trials Regulation (536/2014), there will be a requirement for results of all primary endpoint and patient relevant secondary endpoints to be reported within 12 months of the end of the study.132 The provision of a protocol template alongside example excerpts from trial protocols will help facilitate protocol developers understand how to write high-quality PRO protocol content and support more complete reporting of results.
In a companion paper,15 we also present tools for patient advocates involved in the co-design of trial protocols or the review of protocols through roles on ethics committees or funding committees with PRO endpoints to further optimise study design and facilitate patient involvement. It is essential that patients’ experiences, perspectives, needs and priorities are captured and meaningfully incorporated. The SPIRIT-PRO Group and regulatory agencies strongly support the early and continued involvement of patients and members of the public in trial design and conduct.133 134
The next steps for the SPIRIT-PRO Initiative are to promote uptake and use of the guidelines and implementation tools and development of ethical guidelines for IRBs and ethics committees. The SPIRIT website (www.spirit-statement.org) and PROlearn, a free resource on the optimal use of PROs in research and routine practice (www.bham.ac.uk/prolearn) provides the latest resources and information on the initiative, including a list of supporters. We invite international stakeholders to assist in the evaluation of the SPIRIT Statement and E&E paper by using the documents and providing feedback to inform future revisions.
SPIRIT-PRO forms part of a growing toolbox to promote the optimal use of PROs in trials including guidance for the selection of measures,61 design (SPIRIT-PRO),10 analysis (SISAQOL),34 reporting (CONSORT-PRO)13 and presentation of results (figure 5).135–137 These tools are currently being disseminated by the PROTEUS Consortium (Patient-Reported Outcomes Tools: Engaging Users & Stakeholders) who aim to partner with key patient, clinician, research and regulatory groups around the world to promote the uptake and use of these methodological tools to optimise the assessment and reporting of PROs in clinical trials (https://more.bham.ac.uk/proteus/). Patient and public involvement in all of these activities can help ensure that PRO selection, study implementation and application is transparent, relevant and acceptable. Consistent with this philosophy, patient partners have been involved in all aspects of the development of the SPIRIT-PRO Extension.10 138 139

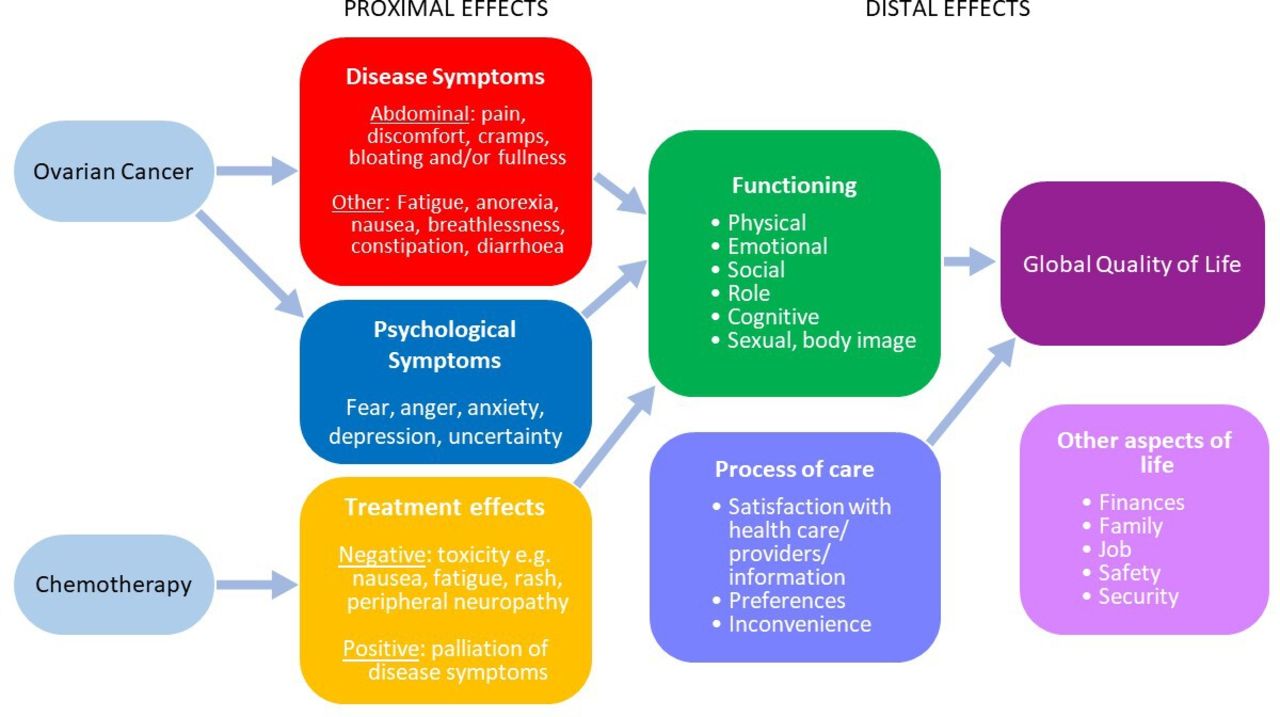{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Resources to promote high-quality patient-reported outcomes (PROs) trial design, analysis and dissemination.9 CONSORT, Consolidated Standards of Reporting Trials; EMA, European Medicines Agency; FDA, Food and Drug Administration; SPIRIT, Standard Protocol Items: Recommendations for Interventional Trials.
Through widespread uptake and support, the potential to improve the completeness and quality of trial protocols and the efficiency of their review can be fully realised. Ultimately, high-quality PRO results can help ensure that important patient-centred evidence on the efficacy, safety and tolerability of interventions is available to inform shared decision-making, labelling claims, clinical guidelines and health policy.
Ethics statements
Ethics approval
Ethical approval was provided by the University of Birmingham Ethical Review Board (ERN_16-0819).
Acknowledgments
The authors would like to thank Trish Groves, MRCPsych. Kluetz P, MD, US Food and Drug Administration, Jeanette Kusel, MSc, National Institute for Health and Care Excellence, Laura-Lee Johnson, PhD, US Food and Drug Administration, Joanna Coast, PhD, University of Bristol and Doug Altman, DSc for their contribution to the SPIRIT-PRO. The SPIRIT-PRO Group gratefully acknowledge the additional contributions as detailed in reference [10], Appendix (refer to online supplemental file 2) made by the SPIRIT-PRO Executive, the ISOQOL Best Practices for PROs in Randomised Clinical Trials Protocol Checklist Taskforce, the international stakeholders responsible for stakeholder survey distribution and stakeholders who completed the stakeholder survey, the Delphi panellists and the SPIRIT-PRO International Consensus Meeting Participants.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @gracemturner
Contributors MC and MK had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. MC and MK are cochairs of the SPIRIT-PRO Group. Concept and design: MC, MK. Acquisition, analysis or interpretation of data: MC, MK, RM-B, OA, DK, AS, A-WC, EB, JBe, ABe, VB, JBl, ABo, JBr, MB, LC, JCC, HD, ACD, CE, LF, RMG, IG, KH, BK-K, LM, SM, TM, LN, JN, DO’C, MP, DP, GP, AReg, ARet, DR, JS, RS, GT, AV, GV, MvH, AW, LW. Drafting of the manuscript: MC, DK, RM-B, AS, A-WC, MK+section writers GV, AReg, DR, ABe, SM, LW, MP, JBr, GT, ARet, AW. Critical revision of the manuscript for important intellectual content: MC, MK, RM-B, OA, DK, AS, A-WC, EB, JBe, ABe, VB, JBl, ABo, JBr, MB, LC, JCC, HD, ACD, CE, LF, RMG, IG, KH, BK-K, LM, SM, TM, LN, JN, DO’C, MP, DP, GP, AReg, ARet, DR, JS, RS, GT, AV, GV, MvH, AW, LW. Obtained funding: MC, DK, MK, Doug Altman, JBl, JBr, MB, Joanna Coast, HD, MvH, J Ives, RM-B, GP, L Roberts, AS. Supervision: MC.
Funding This SPIRIT-PRO Extension was funded by Macmillan Cancer Support (grant 5592105) and the University of Birmingham and was sponsored by the University of Birmingham. Development of the PRO protocol template was funded by an unrestricted educational research grant from UCB Pharma. MC receives funding from the National Institute for Health Research (NIHR) Birmingham Biomedical Research Centre, the NIHR Surgical Reconstruction and Microbiology Research Centre and NIHR ARC West Midlands at the at the University of Birmingham and University Hospitals Birmingham NHS Foundation Trust, Health Data Research UK, Innovate UK (part of UK Research and Innovation), Macmillan Cancer Support, UCB Pharma. MK is supported by the Australian government through Cancer Australia. RM-B is supported by the Australian Government by a National Health and Medical Research Council (NHMRC) research fellowship. JBl is supported by the NIHR Biomedical Research Centre at the University Hospitals Bristol NHS Foundation Trust. She is also an NIHR Senior Investigator. Work was funded by an unrestricted grant from UCB Pharma, and a participant (TM) contributed as a coauthor and member of the industry advisory group.
Disclaimer The study funders/sponsors had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript or decision to submit the manuscript for publication.
Competing interests All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest and declare: SPIRIT-PRO group members were reimbursed for travel/subsistence at the consensus meeting. MC is Director of the Birmingham Health Partners Centre for Regulatory Science and Innovation, Director of the Centre for the Centre for Patient Reported Outcomes Research and is a National Institute for Health Research (NIHR) Senior Investigator. MC has received personal fees from Astellas, Takeda, Merck, Daiichi Sankyo, Glaukos, GSK and the Patient-Centred Outcomes Research Institute (PCORI) outside the submitted work. RM-B reports non-financial support from University of Birmingham. OA, DK and ARet report grants from NIHR. OA and DK report grants from Birmingham Biomedical Research Centre (BRC). OA reports grants from UCB Pharma and also receives funding from the Health Foundation and declares personal fees from Gilead Sciences Ltd. DK and ARet report grants from Innovate UK and Macmillan Cancer Support. DK reports grants from Kidney Research UK, NIHR SRMRC at the University of Birmingham and University Hospitals Birmingham NHS Foundation Trust, personal fees from Merck, GSK. EB declares personal fees from Navigating Cancer, Sivan Healthcare, CareVive Systems and AstraZeneca. Bell reports other from AstraZeneca, is an employee with stock ownership and/or stock options in the company. JCC reports other from Pfizer Inc., and is an employee and a stockholder of Pfizer Inc. IG is a fully paid employee of Boehringer Ingelheim International GmbH. CE is Chair of the Government of Canada Interagency Advisory Panel on Research Ethics. LM reports non-financial support from Daiichi-Sankyo and Cell & Gene Therapy Catapult. Morel reports other from UCB. LN reports other from GlaxoSmithKline, including employment and ownership of stock in GSK. RS reports personal fees from BioMed Central and Pfizer, other from NHS England, NHSx, NDC, NCRI, NIHR, MRC CTU, GeL, Glasgow CTU, UCLH, LSHTM, Cancer Research UK, Macmillan, Warwick University, Warwick CTU and University of Birmingham. AW reports grants from NIHR and Innovate UK. All other authors have completed the ICMJE uniform disclosure form and declare no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous 3 years, no other relationships or activities that could appear to have influenced the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.