





Abstract
Pathogenic viruses represent a small fraction of viral diversity, and emerging diseases are frequently the result of cross-species transmissions. Therefore, we need to develop high-throughput techniques to investigate a broader range of viral biodiversity across a greater number of species. This is especially important in the context of new practices in agriculture that have arisen to tackle the challenges of global food security, including the rising number of marine and freshwater species that are used in aquaculture. In this study, we demonstrate the utility of combining evolutionary approaches with bioinformatics to mine non-viral genome data for viruses, by adapting methods from paleovirology. We report the discovery of a new lineage of dsDNA viruses that are associated with at least fifteen different species of fish. This approach also enabled us to simultaneously identify sequences that likely represent endogenous viral elements, which we experimentally confirmed in commercial salmon samples. Moreover, genomic analysis revealed that the endogenous sequences have co-opted PiggyBac-like transposable elements, possibly as a mechanism of intragenomic proliferation. The identification of novel viruses from genome data shows that our approach has applications in genomics, virology, and the development of best practices for aquaculture and farming.
1. Introduction
Herpesviruses are a large, diverse group of dsDNA viruses that infect humans and other vertebrates. Herpesviral disease can range in severity from mild blisters to cancer, and while their host range is typically narrow, they have the potential to sometimes cross species boundaries. One of the three families in the order Herpesvirales is the Alloherpesviridae that infect amphibians and fish. Despite the critical importance of fish as aquaculture species, only a handful of herpesviruses have been identified, all of which have been responsible for major disease outbreaks that are exacerbated by a poor understanding of fish virology and lack of vaccines against these viruses (Walker and Winton 2010; Murray 2013; Lafferty et al. 2015). For example in the late-90s, a devastating outbreak of Koi herpesvirus rapidly spread to 90% of Israeli carp farms in only a few years resulting in an annual estimated loss of three million USD to the aquaculture industry (Perelberg et al. 2003). Such sudden outbreaks pose an additional risk in that they can threaten wild populations (Walker and Winton 2010; Murray 2013). The development of technologies for sustainable aquaculture will play a crucial role in addressing the challenge of global food security, and this includes monitoring the threat of viral disease.
Viral identification in general, has traditionally involved low throughput techniques such as filtration, microscopy and cell culture. Indeed over the last 50 years, the development of viable fish cell lines has been an invaluable tool in the identification of viral disease agents (Crane and Hyatt 2011). However, such techniques introduce severe biases, including the fact that viruses larger than porcelain filter pores were not known to exist until 2003 (Scola et al. 2003). Moreover, this approach is limited to clinical samples, prohibiting the discovery of potential viral reservoirs in asymptomatic individuals. Such observations, along with important milestones like the discovery of giant viruses, are indicative of a wider need to develop novel approaches to viral discovery in the genomic age (Roux et al. 2015). This includes recognizing the important ecosystem roles of viruses (Suttle 2007), which drives the need to study (and therefore sequence) the total viral content of environmental samples. Moreover, the demand for high throughput techniques also came in the form of ambitious exploratory projects such as mapping the human virome (Delwart 2013). The introduction of next generation sequencing (NGS) has facilitated the rise of culture-independent metagenomic techniques that are unencumbered by the lack of universal genetic markers, and immune to many of the biases in older approaches (Mokili et al. 2012). These techniques are demonstrating their capacity to identify novel viruses in a variety of genomic (Roux et al. 2015; Maumus and Blanc 2016) and transcriptomic databases (Nibert et al. 2016).
NGS has also resulted in the democratization of genome data, which has in turn lead to the establishment of paleovirology, a field that studies the remnants of viruses that have integrated into the genomes of their hosts. Although far more viruses are identified through metagenomics (Edwards and Rohwer 2005), the systematic investigation of host genomes in paleovirology has revealed many novel viral lineages that often represent long extinct viruses (Katzourakis et al. 2007; Belyi et al. 2010a,b; Gilbert and Feschotte 2010; Katzourakis and Gifford 2010; Taylor et al. 2010; Aswad and Katzourakis 2014). This helps to identify ancient groups that could have contemporary relatives, and while most endogenous viral elements (EVEs) are non-functional ‘fossils’ of viruses past, there is increasing recognition of their active influence on host genomes (Mi et al. 2000; Aswad and Katzourakis 2012; Feschotte and Gilbert 2012). However, while the last decade has seen a string of paleovirology milestones with widespread biological significance, nearly all have been limited to mammals, plants and insects (Katzourakis et al. 2009; Katzourakis and Gifford 2010; Horie and Tomonaga 2011; Herniou et al. 2013; Aswad and Katzourakis 2014; Chu et al. 2014; Chuong et al. 2016).
It has previously been suggested that the deployment of a paleovirological approach to viral discovery could be particularly useful in the identification of novel herpesviruses (Houldcroft and Breuer 2015). This proposal supports the findings of our previous study that simultaneously identified paleoviral data and novel herpesviruses among primate host genomes (Aswad and Katzourakis 2014). This raised the intriguing prospect of reapplying our approach to investigate unexplored areas of herpesvirus diversity, where the identification of novel viruses is the main goal rather than a secondary result. Compared to the hundreds of known mammalian herpesviruses, fewer than twenty highly diverse members of the fish-infecting Alloherpesviridae have been identified thus far (twelve are recognized in the latest taxonomy release by the International Committee on Taxonomy of Viruses) (Davison et al. 2009). As well as poor sampling within Alloherpesviridae, the relationship to other large dsDNA viruses is poorly understood. Although they are officially classified as part of the order Herpesvirales, there is limited evidence to support a common origin with other families (McGeoch et al. 2008). Moreover, all of these alloherpesviruses have associated diseases that are known to infect at least a dozen species, often with high rates of mortality (Hanson et al. 2011). However, due to the tissue specificity of fish herpesviruses, many are difficult to culture (Hanson et al. 2011) and indeed there are suspected herpesviruses that have been observed by electron microscopy for decades (Leibovitz and Lebouitz 1985; Shchelkunov et al. 1992; Jakob et al. 2010; Kent and Myers 2000) but unconfirmed through molecular methods. Furthermore, the known herpesviruses that have been identified are limited to widely farmed and well-studied bony fish species (Hanson et al. 2011), which renders future aquaculture developments vulnerable to emerging infection.
Additional data in the form of new viruses, will therefore also be useful in the elucidation of relationships between families in the Herpesvirales order, but also among major lineages of dsDNA viruses more generally. Moreover, by identifying viruses before major disease outbreaks, it could be possible to limit the economic and environmental impact of future epidemics. In this study, we set out to tackle this sampling deficit by repurposing techniques from paleovirology to describe novel viruses. By mining fish genomes for similarity to alloherpesviruses we uncovered virus-like sequences among the genome records of fifteen species of fish from nine different teleost families. These viral sequences share phylogenetic and genomic similarity to alloherpesviruses, and include among them a full-length viral genome associated with the Atlantic salmon (Salmo salar). The preliminary characterization of these viruses also indicates genetic similarity to genes from at least five other viral families, as well as dozens of predicted open reading frames (ORFs) with no detectable similarity to any known protein-coding genes. Finally, there is also evidence to suggest that at least some of the sequences represent EVEs that have heritably integrated into the host genome. In addition to recognizing the importance of identifying these viral sequences specifically, we also conclude that the results strongly advocate in favor of broader and wider scale genomic database mining.
2. Results
2.1 Database mining and phylogenetic reconstruction
Virus-like sequences were identified in 15 different species in a search of 54 teleost fish genomes for similarity to the DNA polymerase of Ictalurid Herpesvirus 1 (IcHV1). Using tBLASTn, we initially considered the top 21 hits that aligned to at least 60% of the query, shortlisting only the top scoring sequences for each species, with the exception of two distinct strains of Oryzias latipes that both contained two distinct hits (Table S1). The average amino-acid identity was 27% between IcHV1 and these 21 hits. We then used 3 sequences from the 11 different species in a BLASTn to reveal similar sequences in an additional 4 species of fish that were an average of 60% identical at the nucleotide level (Table S1). After aligning the shortlisted contigs and delineating the DNA polymerase gene, we performed a BLASTn search against each individual fish genome to retrieve all contigs in the database that contain the DNA polymerase sequence under investigation (Table S2).
Phylogenetic reconstruction revealed that the sequences discovered in fish genome records are a monophyletic group related to other dsDNA viruses. In order to maximize taxonomic sampling, our phylogenetic strategy involved using a short alignment of a conserved region of DNA polymerase. It should be noted that this tree is not an appropriate approach to reconstruct the relationships among large DNA viruses in general. However, based on midpoint rooting, the fish sequences are most closely related to the Alloherpesviridae, with a posterior probability of 100 (Fig. 1A). Posterior probabilities throughout the paper are expressed as values out of 100. Considering the patristic distances among Alloherpesviridae, the sequences are unlikely to be part of the same lineage, but rather represent a distinct clade of previously undescribed viruses. We also investigated the relationships within this clade, using a longer alignment that only considered the new sequences (Fig. 1B). This is because the alignment used to estimate the tree in Fig. 1A was limited to a small conserved region (in order to maximize taxon sample diversity) and does not offer statistically robust information on the relationships among these sequences. In contrast, the tree reconstructed from a longer region of DNA polymerase offers robust support for all nodes in the tree (Fig. 1B). Unlike other dsDNA viruses such as the Herpesviridae, the topology revealed is only somewhat congruent to that of their hosts. For example, the sequences from Salmonidae species and Cichlidae are both monophyletic groups, although the latter clade also contains sequences identified in genomes of Gobidae and Cyprinodontiformes fish (Fig. 1B). On the other hand, there is a broad topological incongruence to the host tree, such as the fact that Salmonidae and Esociformes fish are basal to the Cichlidae (Near et al. 2012), but this is not the case with their associated viral sequences (Fig. 1B).
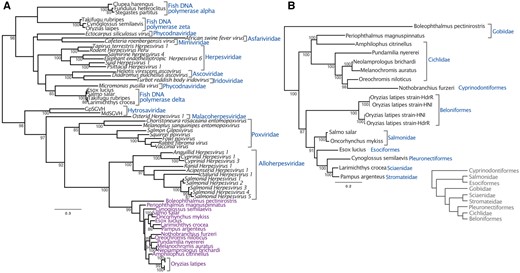
Both panels depict midpoint rooted Bayesian phylogenetic trees reconstructed from an alignment of DNA polymerase. The branch lengths represent the number of substitutions per site and the numbers at each node represent posterior probabilities >85. (A) Posterior probabilities are expressed as values out of 100. As well as the sequences under investigation (annotated in purple) the 233 amino acid alignment included viruses representing eight dsDNA virus families, as well as delta, zeta and epsilon fish DNA polymerases. (B) An extended 2,904-nucleotide alignment of the new sequences without other viral groups intended to obtain more robust support for the topology within the clade. The clades are annotated according to the fish species in whose genome the viral-like data was identified. The inset cladogram shows the relationships between these fish hosts, drawn manually based on the phylogeny in Near et al. (2012).
2.2 Comparative analysis and sequence characterization
The synteny among viral-like DNA polymerase-containing contigs identified in all fifteen species of fish was investigated using the genome aligner MAUVE (Darling et al. 2004) (Fig. 2). Although the length of the contigs found varied greatly, the analysis revealed sizable co-linear blocks among the viral sequences, particularly near DNA polymerase. However, the MAUVE alignment also showed that the contigs are highly rearranged relative to one another, and the length of co-linear blocks is highly variable. We also identified repetitive elements using RepeatMasker 4 in all but the four smallest contigs (Smit et al.). In the case of Esox Lucius we found a pair of contigs that did not contain the same profile of repetitive elements (Fig. 2). This suggests that there are multiple integrations of the viral-like sequences in the E. Lucius genome, supported further by the fact that the sequences do not exhibit collinear blocks. Conversely, two Larimichthys crocea contigs that exhibit 99% nucleotide identity and co-linearity differ by the presence of a LINE (Fig. 2). This could either mean there is more than one locus that recently diverged, or that it reflects heterozygosity of the integrated viral sequence. More generally, the results of the RepeatMasker analysis reveal an abundance of DNA elements, most notably of the Mariner and Mavericks/Polinton type.
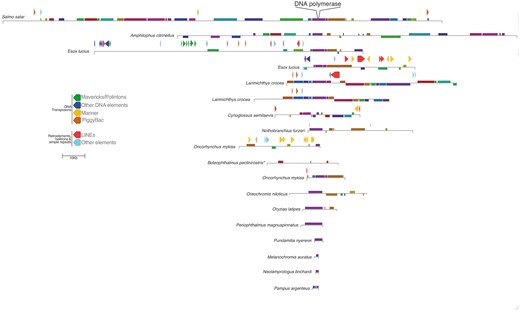
A schematic diagram depicting the blocks of co-linear sequence similarity among the viral contigs, which are drawn relative to the Salmo salar sequence. Homologous blocks across the different sequences are indicated by the same color, and shown below the line if a co-linear block is found in reverse orientation. The alignment is centered at the midpoint of DNA polymerase, which is located in slightly different places for each sequence within the co-linear block. Repetitive elements >200 bp are indicated as arrow blocks above the representation of each contig (excluding simple repeats). *In the case of Boleophthalmus pectinirostris, MAUVE was unable to identify the collinear block containing the DNA polymerase ORFs due to the presence of large insertions not found in other contigs.
ORF prediction using GeneMarkS was performed on the DNA polymerase-containing contigs from four species with the largest contigs (one each from Salmoniformes, and Cyprinodontiformes and two Perciformes) (Fig. 3 and Tables S3–S6). A GenBank formatted file of the predicted ORFs is also provided for convenience for future studies (downloadable from www.paleovirology.com). Consistent with the phylogenetic findings, the majority of predicted ORFs are most similar to genes in members of Alloherpesviridae. However, we also identified ORFs with similarity to other dsDNA viral groups, with irridoviral genes being the second most common top hit (Fig. 3). Across the four annotated contigs, the ORFs are 1,165 bp long on an average with a median length of 834 nt. The longest ORF is 5,916 bp, although no similarity could be detected to any known gene product using BLAST, which was also the case for many of the predicted ORFs. The longest recognizable ORFs were the DNA polymerase-like sequences in Nothobranchius furzeri and Amphilophus citrinellus (4,923 nt and 4,845 nt, respectively). The contig with the highest number of predicted genes (115) was identified in the Salmo salar genome record, which also contains a flanking pair of inverted repetitive regions (∼11 and 5.7 kb) (Fig. 3). This genomic structure is also observed in herpesviruses, which are also between 100 and 200 kb, suggesting that the contig represents a near-complete viral genome.
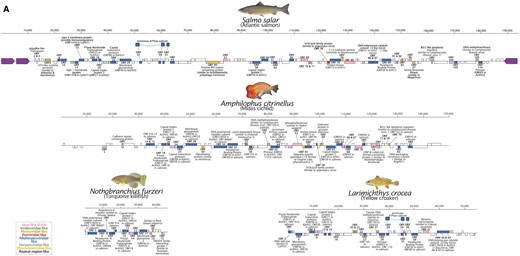
A selection of four of the viral sequences detected in fish genomes are represented here with detailed annotation with predicted open reading frames (ORFs) represented as boxes. ORFs in the forward and reverse orientation are indicated above and below the line, respectively. ORFs without any detectable similarity to known proteins are not labeled, and those with similarity to unnamed proteins are only indicated by their ORF ID. The color-coded key indicates the viral family of the best hit for each predicted ORF. ORFs are completely filled with the corresponding color according to the taxonomic group of the most similar protein, but BLAST similarity was always partial.
We next chose to investigate the Salmo salar results further due to its economic importance and given the fact that there was a high-coverage final assembly available. Interestingly, the large intact contig (AGKD01000001.1, Fig. 3) we identified from the initial assembly was no longer available in the latest version of the sequencing project (but still accessible as an obsolete record). A BLASTn search of this contig against the final assembly revealed that large portions (up to 9 kb) of the viral genome (excluding the repetitive termini) could be found in the majority of salmon chromosomes (Fig. 4). This finding explains the ‘missing contig’ if it was difficult for the assembly algorithm to reconstruct the full-length viral genome once strong evidence for parts of the sequence were found across multiple sites in the salmon chromosomes. To test this rationale further, we designed two confirmatory PCR experiments to check the validity of the multiple-loci result and to confirm that the AGKD01000001.1 contig is representative of a true sequence. In addition to this, we re-mapped the raw sequencing reads against the original contig to search for obvious signs of false assembly (Fig. 4).
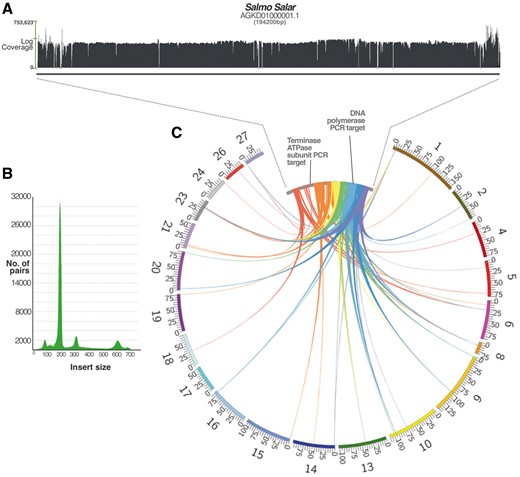
(A) A coverage graph across the length of the salmon contig. The x-axis represents the log coverage, with a horizontal bar indicating the mean at ∼3,000×. Short regions of zero coverage are indicated by a dash along the x-axis, all of which are bridged by read pairs indicating that the contig is not erroneously assembled. (B) The graph shown indicates the number of read pairs for each insert size. The peaks at 180, 300 and 600 correspond to the known sizes of libraries used in the sequencing project. Only high quality, well-aligned and paired reads were included in the coverage count. (C) The figure depicts BLASTn hits of contig AGKD01000001.1 (inner ring) against salmon chromosomes (outer ring), showing only those over 1 kb long (and up to 9 kb) and excluding hits to the highly repetitive terminal ends. All hits are between 80% and 100% identical at the nucleotide level. The colours represent segments of the query sequence AGKD01000001.1 for clarity. The salmon chromosomes are drawn to scale with the values at tick marks representing Mb. AGKD01000001.1 is drawn much larger as it is only 194,200 bp long and would not be visible at the chromosomal scale.
The bioinformatic reconstruction of the AGKD01000001.1 contig revealed strong evidence against miss-assembly. Even if some of the reads belonging to the hypothesized fragments elsewhere in the genome erroneously mapped to the large contig, this is mitigated by the extremely high coverage of paired-end reads of different insert sizes (Fig. 4). About 101 separate libraries were re-mapped to the contig to produce a mean coverage of ∼3000×. We also amplified and sequenced a region from the viral sequence that appeared integrated in multiple locations (a 469-bp region of DNA polymerase). As expected, the sequencing trace files revealed that the DNA polymerase results contained multiple amplicons, as evidenced by several ‘double peaks’ that reflect polymorphic sites between different loci.
We then targeted a region that is only represented in the AGKD01000001.1 contig and other unplaced genomic scaffolds (a 412-bp stretch within ATPase). As predicted, the trace file of this amplicon was indicative of a single sequence, with no evidence of alternative products. These experiments were performed on samples from the same fish used in the genome project (kindly provided by Sigbjørn Lien), as well as additional samples of farmed salmon obtained from local supermarkets and a sushi restaurant.
Given that these results indicate widespread genomic integration of these sequences, we examined the data in search of evidence that might reveal the process of integration. The RepeatMsker results for the AGKD01000001.1 contig revealed the presence of PiggyBac-like transposons at either end of the contig, at the junction between the terminal repetitive region and genic region (Figs 2 and 3). This is consistent with the findings from BLASTp of the predicted ORFs, which identified ORF 2 and 4 as well as ORF 114 and 115 as similar to PiggyBac-derived proteins in fish species. Upon closer examination, we found that these PiggyBac elements are 97% identical to one another. Ordinarily, PiggyBac elements are flanked by terminal inverted repeats (TIR) and ‘TTAA’ motifs. In AGKD01000001.1, rather than each element being flanked by these features, the TIRs and TTAA motif flank the 5′ end of the element on the left side and 3′ end of the element on the right end of the contig (Fig. 5). This suggests that they do not represent separate integrations of two PiggyBac elements.
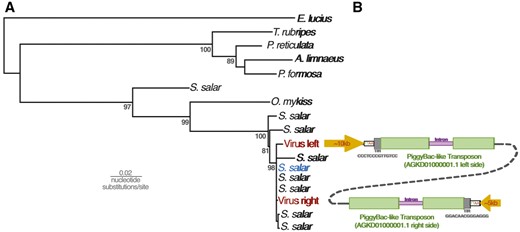
(A) Maximum likelihood phylogenetic tree reconstructed from a 953 nucleotide alignment of a conserved region of PiggyBac-like genes in fish genomes. Numbers at nodes represent percentage results of non-parametric bootstrapping with 1,000 replicates. (B) The PiggyBac-like elements identified in the salmon virus-like contig AGKD01000001.1 are stylistically showing the major genomic features, including the characteristic TTAA motif flanking the elements and a 13-bp terminal inverted repeat. One 750-bp intron is shown in purple, but we cannot rule out the presence of others. The schematic diagrams are not drawn to scale.
Unusually, both PiggyBac-like elements in AGKD01000001.1 contain insertions that disrupt the transposase ORF. A BLAST search against the salmon genome reveals that a large 751-bp ‘insertion’ corresponds to the intron of a PiggyBac-derived gene. Through RNA sequencing and gene prediction analysis, 75 such PiggyBac-like elements have been annotated in the salmon genome. To examine the relationship between our PiggyBac-like elements and those in the salmon genome, we reconstructed a phylogenetic tree including the nine most similar loci from salmon and one representative from the six most similar elements in other fish (Fig. 5). This revealed that both elements are almost identical to a cluster of 5 salmon PiggyBac-derived genes that appear to have diverged from one another relatively recently. However, one of the loci in particular shared the same 750-bp intron, as well as being the only one to exhibit similarity across the whole length of the elements in AGKD01000001.1 (Fig. 5).
3. Discussion
The results of this investigation reveal viral-like sequences identified in open-access genome data of fifteen species of teleost fish. Through phylogenetic reconstruction, we show that this new group is related to alloherpesviruses (either representing a sublineage or new family) and other large dsDNA viruses. Our results include a large contig identified in the S. salar genome data that appears to represent a full-length viral genome, as well as multiple loci within the salmon genome itself. These data will be instrumental for the future characterization of this unusual clade of viruses, several of which are associated with fish that have substantial conservation, research and commercial interest. Moreover, at least two of the viruses we identified are associated with fish that are infected by yet unknown herpesvirus-like pathogens. Salmosalar is affected by papillomatosis, where the fish are affected by lesions of various size, and from which herpesvirus-like particles have been observed (but not in healthy skin) (Shchelkunov et al. 1992). Epidermal hyperplasia has been observed in Esox Lucius, the northern pike, manifesting as flat bluish-white legions, containing Herpesvirus-like capsid structures within an enlarged nucleus (Yamamoto et al. 1984).
It should be noted that the characterization of these sequences as most closely related to alloherpesviruses would be invalidated if the true root of the tree is at the branch leading to the new clade, to the alloherepsviruses or between the two. We opted to root the tree at the midpoint as a conservative solution to the fact that the phylogenetic tree of large dsDNA viruses is extremely difficult to reconstruct with accuracy, due to the complex history of their core genes (e.g. gene loss/gain) (Yutin and Koonin 2012). Moreover, it is not clear what the evolutionary relationship of large dsDNA viral polymerase is with similar hosts genes, which could be due to a number of independent acquisition events (Iyer et al. 2001). Nonetheless, we think that the alloherpesviruses represent the most likely sister group, since our preliminary genomic characterization shows that most predicted ORFs are most similar to alloherpesvirus genes (Fig. 3).
The results are inconclusive as to whether the sequences represent a new family, but there is abundant evidence to suggest that they are not simply alloherpesviruses. First, there is a large patristic distance between the identified sequences and alloherpesviruses, which is much greater than the distances between Alloherpesviridae members. This could either mean that they are a new family or a highly divergent sublineage. Moreover, most of the similarity to alloherpesvirus genes was only partial and shared a low sequence identity. For example, in the S. salar contig, the average identity to alloherpesvirus proteins is 37%, with average gene coverage of 65% (Table S1). Furthermore, as well as similarity to other viral groups (nine ORFs were similar to non-alloherpesviruses), 64/115 of the predicted S. salar ORFs did not exhibit BLAST similarity to known genes.
The topology of the tree without other viral groups indicates that a cross-species transmission of these viruses may have occurred between Periophthalmus magnuspinnatus and Amphilophus citrinellus. This is because the viral sequences identified in these fishes’ genomes form a monophyletic group with strong support despite being associated with different families of fish, although we cannot determine the direction of transfer (Fig. 1B). Similarly, the tree topology suggests that the virus may have transmitted from cichlids to the African killifish Nothobranchius furzeri, which is placed among a clade of sequences associated with cichlid species. More specifically, it is tempting to speculate that the transmission event occurred from the Nile tilapia Oreochromis niloticus, since the two sequences group with posterior probability of 98. Both species are cultivated by humans for food and as a research model (tilapia and killifish, respectively), which could have offered more opportunity for transmission. The tree of these fish sequences also revealed that the two pairs of sequences identified in two Oryzias latipes strains do not group together, suggesting that each genome contains two different strains of the virus (Fig. 1B).
Large-scale metagenomic studies have resulted in an unprecedented rate of viral discovery, but have focused on clinical and environmental samples collected for this purpose (Edwards and Rohwer 2005; Kristensen et al. 2010; Rosario and Breitbart 2011; Mokili et al. 2012). In this paper, we repurposed host genome databases by mining them collectively as a virtual metagenome, adding to a growing trend in searching for viruses in this way (Roux et al. 2015; Maumus and Blanc 2016; Nibert et al. 2016). This approach arises from the recognition that paleovirology and viral metagenomics share an overlapping set of scientific goals along with interchangeable bioinformatic tools (Aswad and Katzourakis 2014), and our results specifically highlight the possibility of identifying sequences that are highly divergent from reference viruses. Once bona fide viral sequences are identified and validated through phylogenetics, we are able to distinguish exogenous viruses from EVEs through a number of telltale signs, such as the presences of in-frame stop codons or frame-shifting indels that typically accumulate through genetic drift at the host neutral rate (Katzourakis and Gifford 2010). Considering the DNA polymerase alignment of over 1,000 amino acids used to reconstruct the fish virus phylogeny (Fig. 1B), frame-shifts were necessary to align the sequences form Esox lucius, Cynoglossus semilaevis, and Boleophthalmus pectinirostris, while in-frame stop codons are found in the Oncorhynchus mykiss and Melanochromis auratus contigs.
In addition to non-sense mutations, a consequence of host genome integration is that mobile elements will integrate into endogenous viral sequences. Among the fish sequences in this study, we uncovered evidence of such elements in the contigs we describe, including many DNA transposons that are closely related to elements identified in other fish (Fig. 2). In particular, the contigs from Esox luicus, Larimichthys crocea, Cynoglussus semilaevis, and Oncorhynchus mykiss include the majority of identified elements, which are mainly either LINEs or belong to the Mariner class of DNA transposons. The latter group of elements are also the most abundant in the salmon genome as a whole (Lien et al. 2016). Moreover, three out of four of these contigs are also the sequences with non-sense mutations in their DNA polymerase gene. Together, this evidence strongly suggests that these sequences are embedded within the genomes of these fish.
Interestingly, although we confirmed that the salmon harbors this viral element at multiple loci in the genome (Fig. 4), the largest contig that we identified resembles a complete viral genome, containing comparatively few hits to mobile elements relative to the contigs from other species (Fig. 2). Apart from two 218-bp regions with similarity to Mariner elements, the only other similarity to transposable elements are hits to two PiggyBac-like transposons at both edges of the genomic region (Fig. 3). Considering that these elements are 97% identical to known salmon PiggyBac-like genes, it is likely that these were recently acquired. PiggyBac elements have been domesticated by their hosts on a number of occasions, and this often involves the appearance of introns (Baudry et al. 2009; Cheng et al. 2010; Pavelitz et al. 2013). A recently described example is PGD5, which is conserved across all vertebrates and in humans is exclusively expressed in neurons (Pavelitz et al. 2013). Our results show that a domesticated salmon PiggyBac gene is associated with the virus-like sequences, which may have facilitated the spread of the element in the salmon genome. Moreover, the repetitive termini of the large contig can be found in the salmon genome, which could have also played a role in the integration of the virus, not unlike the use of telomeric repeats by herpesviruses such as HHV6 and Marek’s Disease Virus (Morissette and Flamand 2010; Greco et al. 2014; Wallaschek et al. 2016).
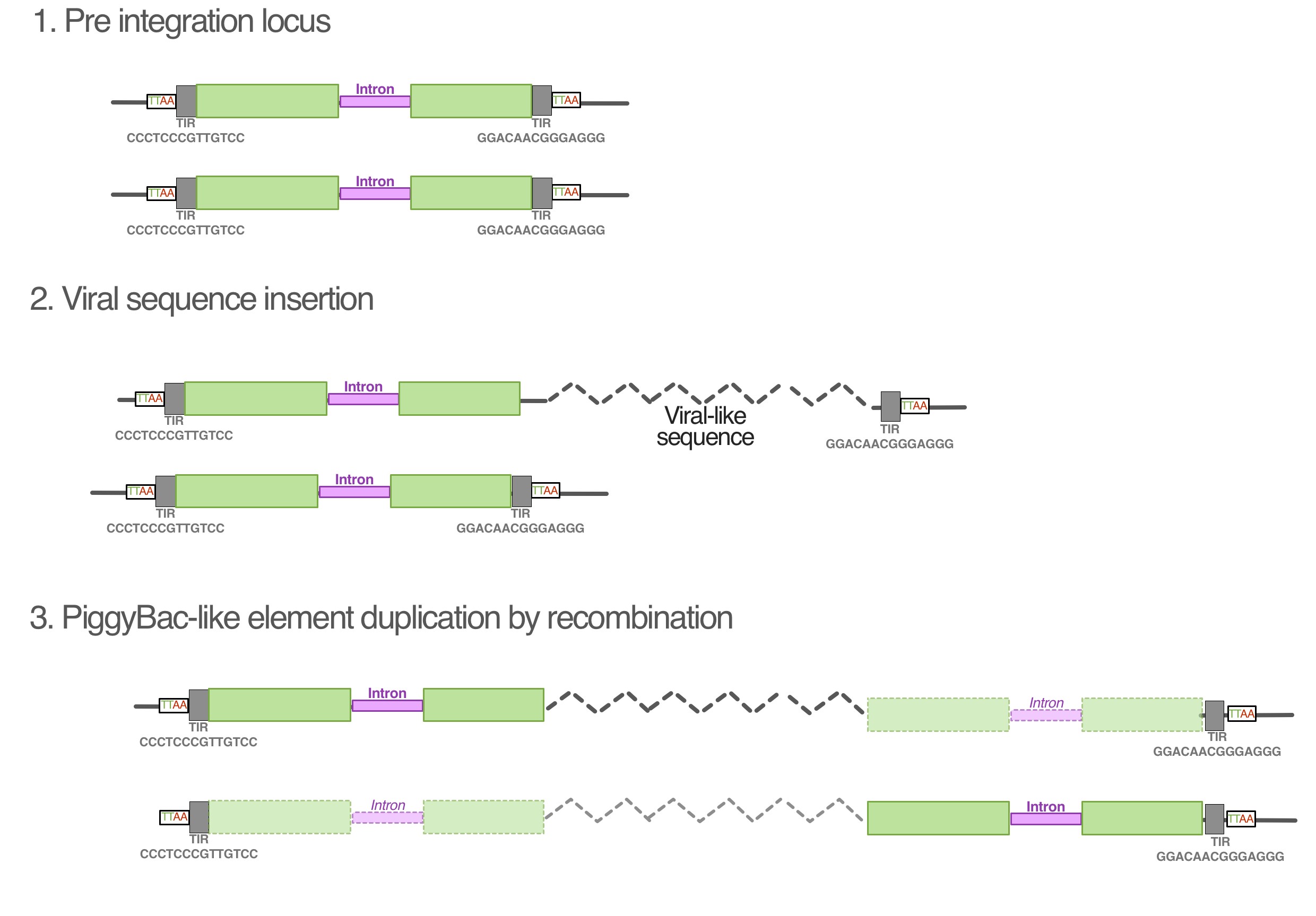
PiggyBac elements transpose via a cut-and-paste mechanism via the encoded transposase that recognizes and binds to short terminal inverted repeats (TIRs) that flank the element. This mechanism is exploited in experimental gene transfer techniques, where TIRs are used in combination with transposase to insert a sequence of choice into a target genome. In the case of AGKD01000001.1, the TIRs flank the entire region that contains recognizable viral genes, which is consistent with arrangement that would be required to transpose the sequence. Unusually, however, the PiggyBac-like elements are found at both ends of the sequences, and each only possesses a single TIR. Our hypothesis for the observed genomic structure is that the viral-like sequences originally integrated into the PiggyBac element upstream of the transposase ORF, but before the 3′ end TIR (Supplementary Fig. S1). This heterozygotic locus could have undergone unequal crossing over with the sister chromatid to duplicate the PiggyBac element on the other side of the virus, resulting in the observed configuration (Supplementary Fig. S1).
Our model is compatible with the observed sequence data, and accounts for the fact that there are two nearly identical PiggyBac-like elements in the same orientation. A recent in-depth analysis into the salmonid-specific whole genome duplication event revealed that the event corresponded to a period of intense transposon activity, possibly due to a compromised transposon regulatory system (Lien et al. 2016). This would have increased the chance of a recombination event with the viral-like sequences, and is consistent with the fact that the PiggyBac-like elements in AGKD01000001.1 are part of a cluster of closely related element sequences. This could also explain the emergence of multiple loci of the virus-like sequence identified in the salmon genome (Fig. 4). However, all of the other loci were partial fragments that could have resulted from the process of salmon genome rediploidization, which involved major genomic rearrangements (Lien et al. 2016). A clear outstanding question that this raises is whether there was a selective process that drove the profile of fragments observed, such as if they provide an evolutionary advantage to the host. This could be in the form of neofunctionalization of the virally derived genes themselves, as has been observed in other hosts that have repurposed such integrations for a variety of functions such as placentation (Mi et al. 2000) or immune defense (Aswad and Katzourakis 2012). Moreover, integrated viral sequences can offer benefits other than gene products, such as regulating adjacent genes, including those that are only a fragment of the original viral insertion (Chuong et al. 2016; Katzourakis and Aswad 2016). In our study, by characterizing the sequences bioinformatically, future endeavors to identify the coding potential of viral insertions will be able to do so in a targeted manner rather than a broad RNA-seq approach.
A potential drawback to relying exclusively on draft assembly data is the certainty with which we can characterize the viral genomes. This is because we have to rely on imperfect genome assembly methods that can erroneously group reads into artificial contigs. However in our study, we have demonstrated that it is possible to achieve a higher level of confidence bioinformatically, through a supplementary investigation into the raw sequencing reads. In the case of the salmon virus, we showed through read mapping that the contig is supported by a mean coverage of ∼3,000×, and there is a mixture of insert-sizes represented that bridge the small number of short ambiguities (Fig. 4). Furthermore, our results are validated by independently verifying the presence of multiple integrated partial viral genomes in several salmon chromosomes through PCR and sequencing, using both the original and unrelated samples.
Innovative sequencing technologies that have enabled high-throughput discovery through metagenomics are beginning to reveal the vastness of unexplored viral biodiversity (Roux et al. 2015; Maumus and Blanc 2016; Nibert et al. 2016). In addition, it has also resulted in the growth of whole genome databases of viral plant and animal hosts, which gave rise to the large-scale exploration of ancient viruses through paleovirology. In the case of these fish viruses, there is great downstream value in discovering novel diversity for epidemiological surveillance and disease management of both wild and farmed populations. Although the results of this analysis undoubtedly require further scrutiny, they exhibit the success of this approach that can be applied to an array of different contexts in basic science and industry.
4. Materials and Methods
4.1 Genome database mining
DNA polymerase protein sequences of four alloherpesviruses and two malacoherpesviruses were used as a query set against the NCBI nr, WGS, HTGS, EST and TSA databases. The search scope was limited to the 54 species of fish for which there are assembled genome sequence data. We retrieved sequences that aligned to at least 60% of the query (ictalurid orf57), choosing a single sequence per species, except for Oryzias latipes, for which there were two different strains. For the second round of searching, we used three representatives from the results of the previous round as search queries, guided by a preliminary phylogeny to avoid choosing redundantly similar sequences. Although all the full-length hits belonged to species identified in the previous round, all three of these searches revealed four additional species that were at least 50% identical (Pampus argenteus, Cynoglossus semilaevis, Melanochromis auratus, and Neolamprologus brichardi). The Salmo salar accession that was eventually used (Table S1) does not correspond to the contig identified through BLAST search in this study. Rather, this contig was first identified in a similar but preliminary search conducted in 2012 against an earlier draft of the genome record.
4.2 Alignment and phylogenetics
The objective of the phylogeny was to determine the relationship of these viral DNA polymerase-like sequences to other viruses. To this end, we aligned representative polymerase sequences from a diverse range of dsDNA virus families, as well as host polymerases that also exhibit similarity to dsDNA virus polymerases. Because of this broad taxonomic strategy, the alignment was limited to the conserved catalytic domain. We used MUSCLE as a preliminary step for alignment, followed by manual adjustment of the positions. For the most divergent sequences, we only aligned the most conserved region of this domain. Columns for which there was no discernable similarity were removed prior to tree reconstruction. We used a Bayesian phylogenetic approach to reconstruct the tree using MrBayes (Ronquist and Huelsenbeck 2003) (10 million generations; 25% burn-in), and implemented the best evolutionary model according to the corrected Akaike Information criterion in ProtTest3 (Darriba et al. 2011) (WAG + G). The alignments are available upon request.
4.3 Contig annotation, synteny mapping and visualization
Similarly to other studies on alloherpesvirus genomes (van Beurden et al. 2010), ORFs were identified using GeneMarkS (Besemer 2001). The output GFF files were converted into GenBank records and visualized using MAUVE (Darling et al. 2004) with further graphical editing in Pixelmator 3.3. MAUVE was also used for a whole contig alignment of all of the species represented in the phylogeny, to identify ‘locally co-linear blocks’ with the Salmo salar contig as the reference. Repetitive elements in each contig were identified using RepeatMasker, excluding the reporting of simple repeats and low-complexity regions. The results were then visualized in IGV and transposed onto the ORF maps. The amino acid sequence for each of the predicted ORFs was used as a query for a BLASTp search against a database of all dsDNA viral proteins in the NCBI nr database. Only results with an e-value better than 1×10−4 were considered, and a table was constructed detailing the results for the top-scoring hit of each ORF. For ORFs that did not exhibit similarity to any dsDNA virus proteins, we performed a second more exhaustive search of the rest of the nr database. Read mapping for salmon contig AGKD01000001.1 was performed using the BWA mem algorithm using the salmon contig as a reference sequence for 101 mate-paired libraries of the salmon genome project (NCBI Accession: PRJNA72713). The mapped reads were strictly filtered for erroneous alignment using BAMtools filter, leaving only those reads that mapped in a proper pair with an edit distance of <5.
4.4 PCR amplification and sequencing
In addition to DNA samples of ‘Sally’ kindly provided by Sigbjørn Lien and colleagues, we extracted DNA from salmon fillet and salmon sashimi using the DNeasy Blood and Tissue kit by Qiagen. The supermarket-bought fish were obtained from Tesco and Sainsbury’s in Oxford, UK. We targeted a 469-bp region with similarity to an alloherpesvirus-like ATPase with the following primers designed in Primer3: F-gctagtaacagcctcatcattaaac R-ccgagtacaatagactgatgaaaga. We also amplified and sequenced a 412 bp of an ORF with similarity to DNA polymerase using the following primers: F-cttcaaggttataacatcgtc R-caggagagtcaagttggag. We used the RedTaq Readymix master mix by Sigma-Aldrich to perform the PCR, which includes all reagents and polymerase. Source Bioscience performed Sanger sequencing of both the forward and reverse strand, and the trace files were viewed in four peaks.
Supplementary data
Supplementary data are available at Virus Evolution online.
Acknowledgements
We would like to thank Pakorn Aiewsakun and Tara Hurst for helpful comments on the manuscript. We are grateful to Sigbjørn Lien, Kristil Kindem Sundsaasen and Odd-Arne Olsen for providing us with the salmon DNA samples.
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}