





Abstract
Erythropoietin (EPO) improves cardiac function and induces neovascularization in chronic heart failure (CHF), although the exact mechanism has not been elucidated. We studied the effects of EPO on homing and incorporation of endothelial progenitor cells (EPC) into the myocardial microvasculature and myocardial expression of angiogenic factors.
CHF was induced in rats by coronary artery ligation resulting in myocardial infarction (MI) after bone marrow had been replaced by human placental alkaline phosphatase (hPAP) transgenic cells. We studied the effects of darbepoetin alfa treatment (EPO, 40 µg/kg, every 3 weeks, starting 3 weeks after MI) on longitudinal changes in left ventricular (LV) function, circulating EPC, myocardial histology, and expression of vascular endothelial growth factor (VEGF) determined 9 weeks after MI. EPO prevented LV-dilatation and improved cardiac function (all P < 0.05), which was associated with 42% increased capillary growth (P < 0.01). EPO-induced mobilization of EPC from the bone marrow (P < 0.01), which resulted in a three-fold increased homing of EPC into the cardiac microvasculature. The percentage of the endothelium that consisted of bone marrow derived cells was significantly increased (3.9 ± 0.5 vs. 11.4 ± 1%, P < 0.001) comprising 30% of the newly formed capillaries. In addition, EPO treatment resulted in a 4.5-fold increased myocardial expression of VEGF, which correlated strongly with neovascularization (r = 0.67; P < 0.001). VEGF was equally expressed by endothelial cells of myocardial and bone marrow origin.
EPO-induced neovascularization in post-MI heart failure is mediated through a combination of EPC recruitment from the bone marrow and increased myocardial expression of VEGF.
Introduction
Chronic heart failure (CHF) represents a complex of symptoms related to impaired cardiac function affecting 5 million people in the US. Despite optimal treatment with current strategies, morbidity and mortality remain high.1
CHF is associated with myocyte hypertrophy and impaired microvascularization of the myocardium, leading to a mismatch between oxygen demand and supply.2,3 Current therapies aimed at improving the microvasculature are under investigation, of which erythropoietin (EPO) is one of the most promising therapies.4–6 EPO is an erythropoietic growth factor, promoting survival, proliferation, and differentiation of erythroid progenitor cells.7 We and others have recently demonstrated that EPO-treatment improved cardiac function in experimental models of CHF.8–12 The improvement of cardiac function is consistently associated with an increase in capillary formation, although the mechanism of neovascularization is unknown. It has repeatedly been shown that EPO promotes proliferation and survival of endothelial cells in vitro and stimulates angiogenesis in vivo.13–15 In addition, EPO induces the proliferation, differentiation, and adhesion of a subset of bone-marrow-derived progenitor cells with an endothelial phenotype [endothelial progenitor cells (EPC)] in vitro and results in marked mobilization of EPC in vivo.8,16–18 EPC specifically home to sites of neovascularization and incorporate into newly formed vessels.19 It is, however, unknown whether the mobilized EPC contribute to EPO-induced neovascularization.
We hypothesized that EPO-induced neovascularization is mediated through EPC recruited from the bone marrow in addition to in situ proliferation of myocardial endothelial cells. In order to study the effects of EPO on differentiation of EPC into the myocardial vasculature, bone marrow of rats was replaced with labelled bone marrow cells.
Methods
Animals
We used male Fischer F344 rats (n = 65) weighing 200–230 g (Charles Rivers, France) as recipients and R26-hPAP rats [n = 10, F344 background, ubiquitously expressing human placental alkaline phosphatase (hPAP)] as donors.20 Animals were fed and housed, according to institutional rules, and the experimental protocol was approved by the Animal Ethical Committee of the University of Groningen.
Bone marrow labelling
In order to evaluate the effects of EPO on homing and incorporation of EPC into the endothelium, we replaced the bone marrow by genetically labelled bone marrow cells as described in detail previously.21 Briefly, 24 h after 9 Gray total body irradiation of recipient rats, whole bone marrow cells (25 × 106 nucleated cells) of donor rats were transfused to recipients. Rats were housed in filtertop cages and drinking water was supplemented with neomycin (0.35% wt/vol), 2 weeks post and prior to irradiation. After 6 weeks, hPAP expression on leucocytes was evaluated by FACS, and rats with chimerism > 80% were randomly allocated to MI or sham surgery. Successful haematopoietic recovery was further confirmed by full blood counts and chimerism was re-evaluated at sacrifice. This model allowed us to evaluate differentiation of bone-marrow-derived cells into an endothelial phenotype.
Experimental protocol
Rats were randomly subjected to induction of myocardial infarction (MI) or sham surgery as described previously in detail.11 Briefly, animals were intubated and mechanically ventilated with 2.5% isoflurane in room air enriched with 1.0 L/min oxygen. After left-side thoracotomy, MI was induced by ligating the proximal portion of the left coronary artery. In sham operated rats, the same surgery was performed without ligating the suture. Three weeks after coronary artery ligation, rats with MI were subjected to treatment with the long-acting EPO analogue darbepoetin alfa (40 µg/kg, Aranesp, Amgen Inc., Thousands Oaks, CA, USA) or saline, administered intraperitoneally once every 3 weeks. Treatment allocation was balanced for left ventricular (LV) end-diastolic diameter and LV fractional shortening determined by echocardiography. The dose of darbepoetin was based on our previous experiments, demonstrating increased neovascularization in this model.11 At baseline, at week 3 (before therapy) and weeks 6 and 8, cardiac function was determined by echocardiography. After 9 weeks, haemodynamic function was assessed invasively, thereafter hearts were rapidly excised and weighed. Myocardial tissue was transected transversally and processed for immunohistochemistry or snap frozen for western blot analysis.
Echocardiographic measurements
Cardiac function was prospectively assessed by echocardiography (Vivid 7, GE Healthcare, Chalfont St Giles, UK; equipped with a 10-MHz phase array linear transducer). The echocardiographic measurements were performed under general anesthesia with 2.5% isoflurane, by two researchers blinded for the treatment allocation. Both 2-dimensional (2D) images in parasternal long-axis and short-axis view and 2-D guided M-mode tracing were obtained. Long-axis views were obtained, ensuring that the mitral and aortic valves and the apex were visualized. Short-axis views were recorded at the level of mid-papillary muscles. LV end-systolic diameter (LVESD) and LV end-diastolic diameter (LVEDD) were measured from the M-mode and calculated as an average from short- and long-axis view. LV fractional shortening (FS %) was calculated as FS = (LVEDD − LVESD)/LVEDD × 100%. LV ejection fraction (EF %) was calculated using the Teichholz method of estimated LV volumes.22
Haemodynamic measurements
At sacrifice, rats were anesthetized and a microtip pressure transducer (Millar Instr. Inc., Houston, TX, USA) was inserted into the LV cavity via the right carotid artery. After a 3-min period of stabilization, heart rate (HR), LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), and developed LV pressure (dLVP = LVSP − LVEDP) were measured. As indices of contractility and relaxation, the maximal rates of increase and decrease in LVP (dP/dtmax and dP/dtmin) were determined. The catheter was retracted into the aortic arch and arterial systolic/diastolic blood pressures (SBP, DBP) were recorded.
Infarct size, myocyte hypertrophy, and capillary density
Infarct size, myocyte hypertrophy, and capillary density were determined as described in detail previously.11 Briefly, infarct size was determined by planimeter in transverse slices on picrosirius red/fast green–stained sections and expressed as the percentage of scar length to total LV circumference. Concentric myocyte hypertrophy, in the viable LV wall remote from the infarct, was measured in deparaffinized sections stained with Gomori's silver staining, as the cross-sectional area of transversally cut myocytes showing a nucleus, and averaged per tissue area (millimetre2). Endothelial cells were stained with biotin-labelled GSL-Lectin (1:100; Sigma-Aldrich, St Louis, MO, USA), a size criterion of 10 µm was used to exclude small arterioles and venules, and image analysis was used to measure capillary density in the viable LV-wall, calculated as the number of capillaries per tissue area (millimetre2) in the all transverse slice. As a measure of neovascularization, capillary-to-myocyte ratio was calculated dividing capillary with myocyte density.
Circulating endothelial progenitor cells
Whole blood was collected in heparine tubes (17 IU/mL). Mononuclear cells were isolated using Histopaque-1083 (Sigma Chemical, St Louis, MO, USA) according to the instructions supplied by the manufacturer. Isolated mononuclear cells (1 × 106) were seeded on fibronectin-precoated 24-well plates (BD BioCoat, Bedford, MA, USA) in EndoCult medium (StemCell Technologies, London, UK) supplemented with Penicillin (100 U/mL) and Streptomycin (100 µg/mL). After 6 days, adherent cells were washed with medium, incubated with 1,1′-dioctadecyl-3,3,3′, 3′-tetramethylindocarbocyanine-labelled acetylated LDL (10 µg/mL Dil AcLDL; Molecular Probes, Invitrogen, Carlsbad, CA, USA) for 12 h, fixed with 1% paraformaldehyde for 10 min, and counterstained with fluorescein isothyiocyanate-labelled Griffonia (bandeiraea) simplicifolia lectin I, isolectin B4 (lectin, 10 µg/mL; Vector Laboratories, Burlingame, CA, USA) and DAPI for nuclear staining. Images were captured by an LSM 410 confocal microscope (Carl Zeiss, Jena, Germany). Cells double positive for DiI AcLDL and lectin staining were considered EPC and counted in high-power fields by co-localization analysis (Image-Pro Plus for Windows, version 4.5.0.29). For every rat, cells were seeded in duplicate and an average number of EPC was calculated per well from five high-power fields.
Fluorescent microscopy
Cryosections were stained with primary antibodies [rabbit anti-hPAP, serotec, London, U.K., mouse anti-rat His52 (rat endothelial cell antigen) a kind gift from Dr J.L. Hillebrands, and mouse anti-vascular endothelial growth factor (VEGF), C-1, Santa Cruz] followed by FITC labelled goat anti rabbit-IgG- and TRITC, Alexa 555 or Cy5-labelled goat anti-mouse-IgG-isotype specific secondary antibodies. For nuclear staining, sections were mounted in vectashield mounting medium containing DAPI. Analysis was restricted to the viable LV wall. Cells that stained positive for hPAP were considered bone-marrow-derived and cells double positive for hPAP and His-52 were considered bone-marrow-derived endothelial cells. Cells were enumerated per high power field by two researchers (B.D.W., H.O.) blinded for the treatment allocation in five randomly chosen high power fields of the non-infarcted LV free wall. The percentage of the endothelium composed of bone-marrow-derived cells was calculated by co-localization analysis. The surface area that stained double positive for hPAP and his52 is expressed as percentage of the total his-52 positive area.
Erythropoietin, erythropoietin-receptor, and vascular endothelial growth factor expression
In order to evaluate the effects of EPO on expression of angiogenetic factors and the EPO receptor, samples of the viable LV free wall (non-infarcted area) were snap frozen in liquid nitrogen and stored at − 80°C. The expression of EPO, EPO-receptor, and VEGF was determined in tissue homogenates of seven randomly selected rats per treatment group by standard western blotting techniques under denaturing conditions, as described previously.23 Membranes were reprobed for GAPDH to confirm equal protein loading and transfer. Signals were detected by the ECL-detection method and quantified by densitometry. Results are expressed as arbitrary units and represent the ratio between EPO, EPO-receptor, or VEGF and GAPDH per lane. Primary antibodies were purchased from Santa Cruz biotechnology [EPO H126, 1:250; EPO-receptor M-20, 1:500; VEGF C1, 1:250 (recognizes all splice variants of VEGF)] or Fitzgerald Industries (GAPDH 6c5, 1:10000) and horeseradish peroxidase conjugated anti-mouse or anti-rabbit IgG were used as secondary antibodies.
Plasma erythropoietin and vascular endothelial growth factor levels
Plasma levels of EPO and VEGF levels were determined by a commercial Enzyme Linked Immunosorbent Assay according to the guidelines provided by the manufacturer (Quantikine, R&D systems, Londen, UK).
Statistical analysis
Data are presented as mean ± SEM when normally distributed and median + interquartile range when skewed distributed. Differences among groups were tested using one-way analysis of variance, followed by LSD post-hoc analysis if normally distributed, and by Kruskal–Wallis test followed by Mann–Whitney U test with Bonferroni correction when skewed. Correlations were assessed with Spearman's correlation test. All reported probability values are two-tailed, and a P-value < 0.05 was considered statistically significant. All statistical analyses were performed with SPSS version 12.0.
Results
Erythropoietin prevents progression of left-ventricular-dysfunction and improves capillary density, associated with marked mobilization of endothelial progenitor cells
Three rats were excluded from analysis due to failed bone marrow transplantation. Twenty-four hour mortality after MI was 31% (16 rats) and three rats (two in MI–EPO and one in MI) were excluded because the infarct size was < 25% of the LV-circumference. The final population comprised 12 rats per MI group and 9 rats in the sham group. General characteristics are presented in Table 1. Although haematocrit values were comparable between groups at the start of treatment, EPO treatment resulted in maximal 31% increase in haematocrit increasing from 50 ± 0.3 to 64 ± 1% 1 week after treatment. Although the haematocrit decreased during the following 2 weeks until the next EPO treatment, it remained significantly elevated in MI–EPO compared with sham and MI throughout the experiment (all P < 0.01).
General characteristics and myocardial function
| Sham | Myocardial infarction | Myocardial-infarction–erythropoietin | |
|---|---|---|---|
| General | |||
| n | 9 | 12 | 12 |
| Infarct size (% of LV)a | — | 43 ± 1 | 41 ± 2 |
| Haemodynamics | |||
| Heart rate (bpm) | 300 ± 17 | 274 ± 11 | 295 ± 8 |
| LVSP (mmHg) | 125 ± 6 | 108 ± 4† | 120 ± 3‡ |
| LVEDP (mmHg) | 13 ± 3 | 22 ± 1† | 16 ± 1‡ |
| dLVP (mmHg) | 112 ± 8 | 86 ± 5† | 103 ± 4‡ |
| SBP (mmHg) | 114 ± 14 | 108 ± 4 | 120 ± 3 |
| DBP (mmHg) | 88 ± 3 | 78 ± 4 | 90 ± 2§ |
| dP/dt max (mmHg/s) | 11847 ± 761 | 9180 ± 537† | 10896 ± 336*‡ |
| dP/dt min (mmHg/s) | −10368 ± 800 | −7311 ± 595† | −8749 ± 287*‡ |
| Body/organ weight | |||
| BW (g) | 293 ± 5 | 281 ± 5 | 290 ± 6 |
| Heart weight/BW (mg/g) | 3.0 ± 0.9 | 4.0 ± 0.7† | 3.6 ± 0.2†‡ |
| Sham | Myocardial infarction | Myocardial-infarction–erythropoietin | |
|---|---|---|---|
| General | |||
| n | 9 | 12 | 12 |
| Infarct size (% of LV)a | — | 43 ± 1 | 41 ± 2 |
| Haemodynamics | |||
| Heart rate (bpm) | 300 ± 17 | 274 ± 11 | 295 ± 8 |
| LVSP (mmHg) | 125 ± 6 | 108 ± 4† | 120 ± 3‡ |
| LVEDP (mmHg) | 13 ± 3 | 22 ± 1† | 16 ± 1‡ |
| dLVP (mmHg) | 112 ± 8 | 86 ± 5† | 103 ± 4‡ |
| SBP (mmHg) | 114 ± 14 | 108 ± 4 | 120 ± 3 |
| DBP (mmHg) | 88 ± 3 | 78 ± 4 | 90 ± 2§ |
| dP/dt max (mmHg/s) | 11847 ± 761 | 9180 ± 537† | 10896 ± 336*‡ |
| dP/dt min (mmHg/s) | −10368 ± 800 | −7311 ± 595† | −8749 ± 287*‡ |
| Body/organ weight | |||
| BW (g) | 293 ± 5 | 281 ± 5 | 290 ± 6 |
| Heart weight/BW (mg/g) | 3.0 ± 0.9 | 4.0 ± 0.7† | 3.6 ± 0.2†‡ |
*P < 0.05.
†P < 0.01 vs. sham.
‡P < 0.05.
§P < 0.01 vs. MI.
Data are presented as mean ± SEM; n indicates number of animals; bpm, beats per minute; LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure; dLVP, developed left ventricular pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; dP/dt max and dP/dt min, maximal rates of increase and decrease in LVP; BW, bodyweight.
aInfarct size, as % of LV-circumference of the mid papillary slice.
General characteristics and myocardial function
| Sham | Myocardial infarction | Myocardial-infarction–erythropoietin | |
|---|---|---|---|
| General | |||
| n | 9 | 12 | 12 |
| Infarct size (% of LV)a | — | 43 ± 1 | 41 ± 2 |
| Haemodynamics | |||
| Heart rate (bpm) | 300 ± 17 | 274 ± 11 | 295 ± 8 |
| LVSP (mmHg) | 125 ± 6 | 108 ± 4† | 120 ± 3‡ |
| LVEDP (mmHg) | 13 ± 3 | 22 ± 1† | 16 ± 1‡ |
| dLVP (mmHg) | 112 ± 8 | 86 ± 5† | 103 ± 4‡ |
| SBP (mmHg) | 114 ± 14 | 108 ± 4 | 120 ± 3 |
| DBP (mmHg) | 88 ± 3 | 78 ± 4 | 90 ± 2§ |
| dP/dt max (mmHg/s) | 11847 ± 761 | 9180 ± 537† | 10896 ± 336*‡ |
| dP/dt min (mmHg/s) | −10368 ± 800 | −7311 ± 595† | −8749 ± 287*‡ |
| Body/organ weight | |||
| BW (g) | 293 ± 5 | 281 ± 5 | 290 ± 6 |
| Heart weight/BW (mg/g) | 3.0 ± 0.9 | 4.0 ± 0.7† | 3.6 ± 0.2†‡ |
| Sham | Myocardial infarction | Myocardial-infarction–erythropoietin | |
|---|---|---|---|
| General | |||
| n | 9 | 12 | 12 |
| Infarct size (% of LV)a | — | 43 ± 1 | 41 ± 2 |
| Haemodynamics | |||
| Heart rate (bpm) | 300 ± 17 | 274 ± 11 | 295 ± 8 |
| LVSP (mmHg) | 125 ± 6 | 108 ± 4† | 120 ± 3‡ |
| LVEDP (mmHg) | 13 ± 3 | 22 ± 1† | 16 ± 1‡ |
| dLVP (mmHg) | 112 ± 8 | 86 ± 5† | 103 ± 4‡ |
| SBP (mmHg) | 114 ± 14 | 108 ± 4 | 120 ± 3 |
| DBP (mmHg) | 88 ± 3 | 78 ± 4 | 90 ± 2§ |
| dP/dt max (mmHg/s) | 11847 ± 761 | 9180 ± 537† | 10896 ± 336*‡ |
| dP/dt min (mmHg/s) | −10368 ± 800 | −7311 ± 595† | −8749 ± 287*‡ |
| Body/organ weight | |||
| BW (g) | 293 ± 5 | 281 ± 5 | 290 ± 6 |
| Heart weight/BW (mg/g) | 3.0 ± 0.9 | 4.0 ± 0.7† | 3.6 ± 0.2†‡ |
*P < 0.05.
†P < 0.01 vs. sham.
‡P < 0.05.
§P < 0.01 vs. MI.
Data are presented as mean ± SEM; n indicates number of animals; bpm, beats per minute; LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure; dLVP, developed left ventricular pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; dP/dt max and dP/dt min, maximal rates of increase and decrease in LVP; BW, bodyweight.
aInfarct size, as % of LV-circumference of the mid papillary slice.
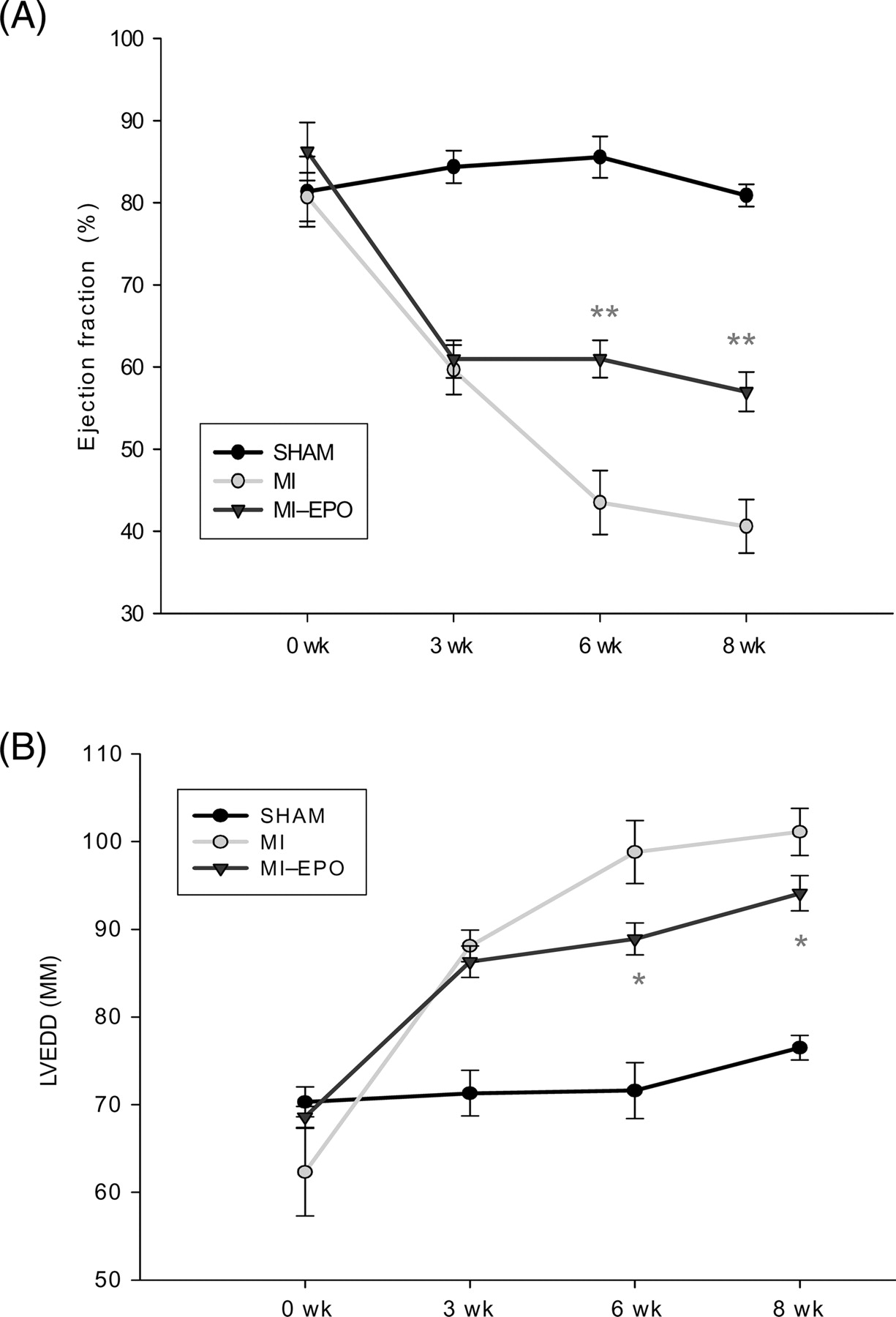
Serial echocardiographic parameters are presented in Figure 1. Induction of MI resulted in marked enlargement of LVEDD and deterioration of LV performance (EF) at week 3. In the following weeks, the deterioration of LV performance and LV dilatation (LVEDD) progressed in the untreated MI group, but remained stable in the EPO treated group (all P < 0.05). LV-systolic pressure, LV developed pressure, myocardial contractility (dP/dtmax) and relaxation (dP/dtmin) and LVEDP were all significantly impaired in both MI groups compared with sham, and significantly improved after EPO treatment (Table 1).
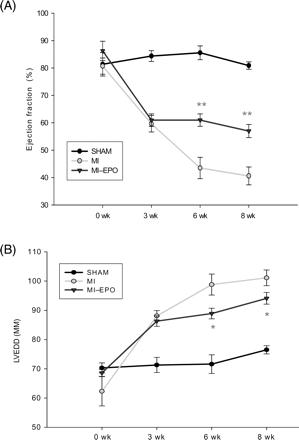
Effect of erythropoietin on myocardial remodelling after myocardial infarction. Changes in echocardiographic indices of left ventricular function and left ventricular end-diastolic diameter during 8-weeks follow-up after coronary artery ligation. *P < 0.05 vs. myocardial infarction, **P < 0.01 vs. myocardial infarction. LVEDD indicates left ventricular end-diastolic diameter.
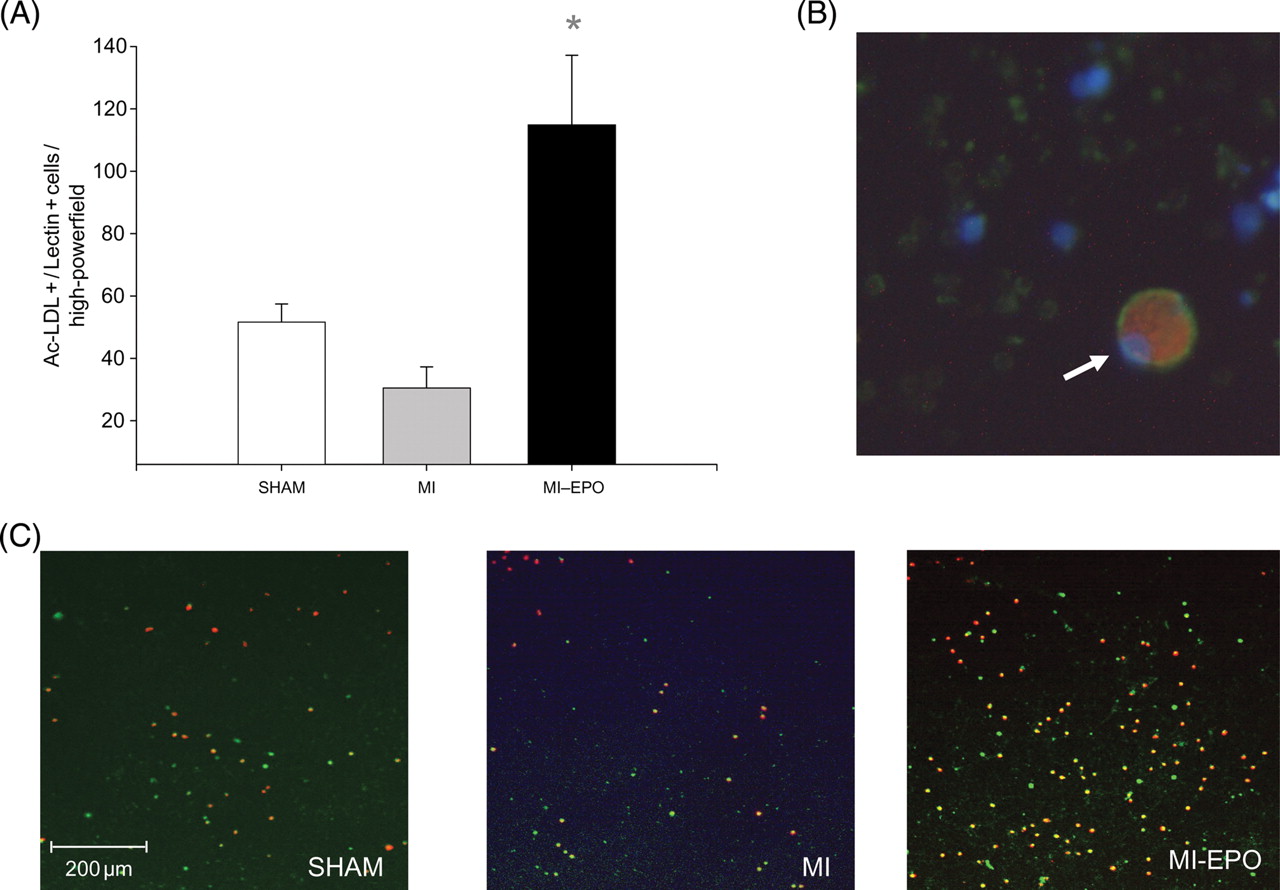
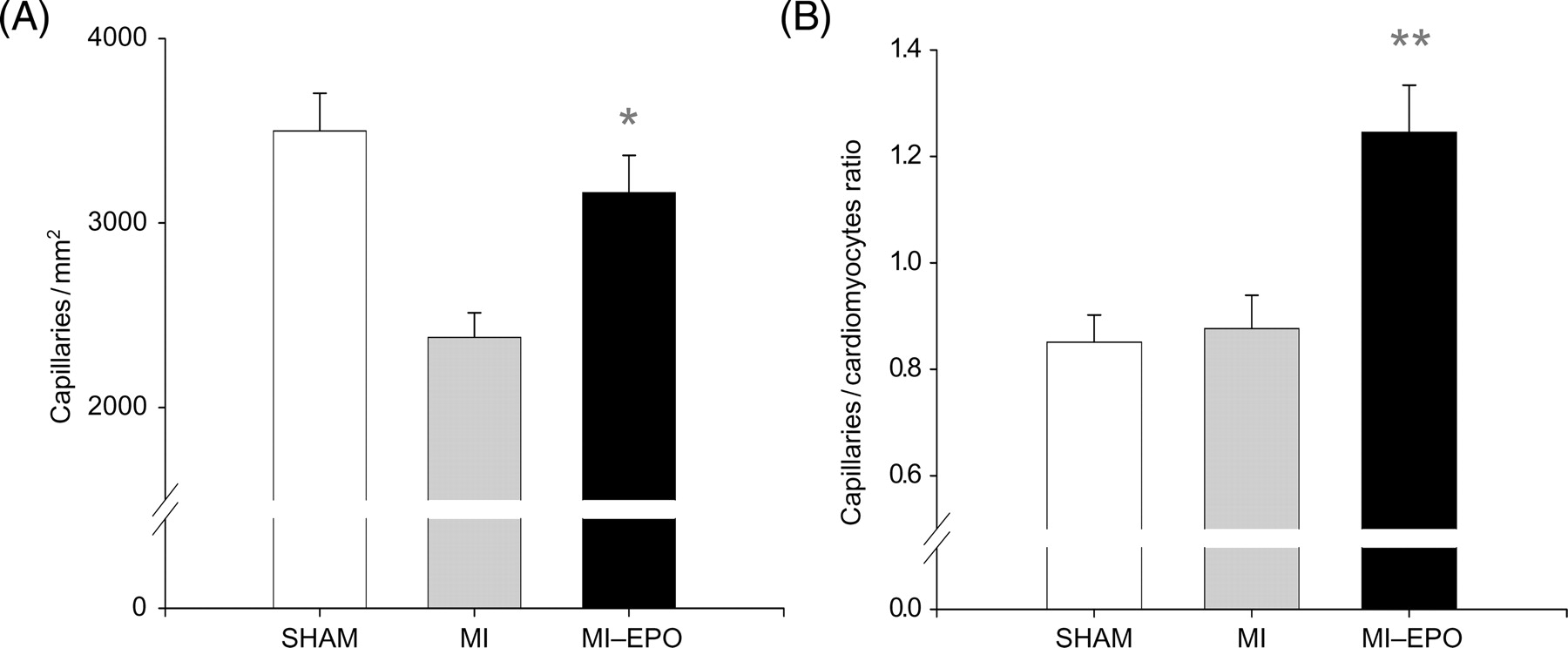
Histological infarct size was comparable between all MI groups. Cardiomyocytes' cross-sectional area increased in both MI groups compared with sham (P < 0.01), but was not significantly different between MI and MI–EPO. Capillary density was significantly reduced in the saline treated MI group (P < 0.01). EPO treatment increased capillary density by 33% (Figure 2, P < 0.01) restoring it to sham levels. The capillary-to-myocyte ratio increased by 42% in the EPO treated group compared with sham and MI (Figure 2, P < 0.01), indicating the formation of new vessels, rather than resulting from reduced cardiomyocyte hypertrophy. Treatment with EPO resulted in a four-fold increase in the number of circulating EPC compared with the MI group (P < 0.01; Figure 3).
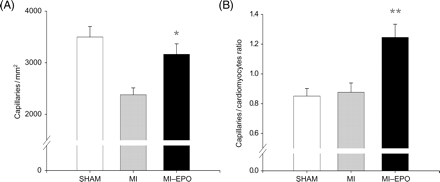
Effect of erythropoietin treatment on neovascularization. (A) Measurements of capillary density in number of capillaries per millimetre2. (B) Bar graph representing the capillary-to-myocyte ratio in different groups. *P < 0.05 vs. myocardial infarction, **P < 0.01 vs. myocardial infarction.
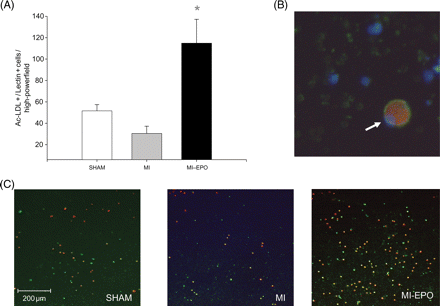
Effects of erythropoietin treatment on number of circulating endothelial progenitor cells. (A) Graphic representation of number of endothelial progenitor cells. (B) Endothelial progenitor cell under high magnification (white arrow), positively stained for DiI AcLDL (red cytoplasm) and lectin (green cytoplasm), including DAPI nuclear staining (blue). (C) Representative microscopic fields from all experimental groups, with double stained (green + red) positive endothelial progenitor cells. *P < 0.01 vs. myocardial infarction.
Erythropoietin augments homing and incorporation of endothelial progenitor cells into the myocardial microvasculature
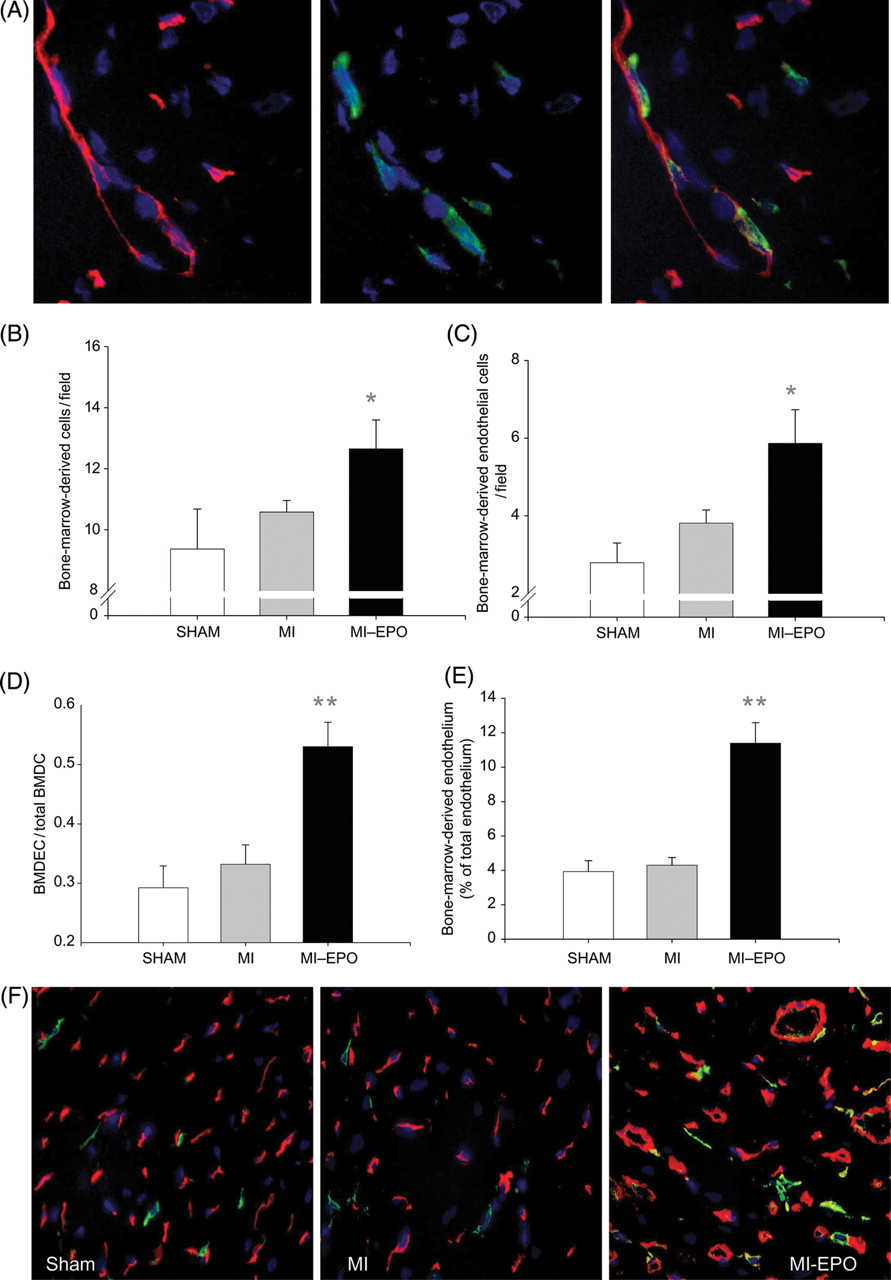
In the non-infarcted LV free wall of the MI rats, a slight non-significant increase in the number of bone-marrow-derived cells (hPAP+) and bone-marrow-derived endothelial cells (hPAP+, his 52+) was observed compared with sham (Figure 4). This was markedly stimulated by EPO treatment, resulting in a 29% increased influx of bone-marrow-derived cells and a 2.1-fold increase in bone-marrow-derived endothelial cells (P < 0.05) The percentage of the endothelium that comprised bone-marrow-derived cells did not differ significantly between MI and sham groups. However, this was markedly augmented by EPO treatment, increasing from 3.9 ± 0.5 to 11.4 ± 1% (Figure 4; P < 0.001 vs. sham and MI). EPO treatment resulted in a 33% increase in capillary density and the percentage of the endothelium that comprised bone-marrow-derived cells increased from 4–11%. Therefore, ∼30% of the newly formed endothelium comprised bone-marrow-derived cells. The enhanced incorporation resulted from specific homing of EPC, reflected by a significantly higher ratio between bone-marrow-derived endothelial cells and total bone-marrow-derived cells (P < 0.001, Figure 4). Moreover, a strong correlation was observed between circulating EPC and myocardial bone-marrow-derived endothelial cells (R = 0.64, P < 0.001) indicating that the increased EPC incorporation was directly related to increased EPC mobilization.
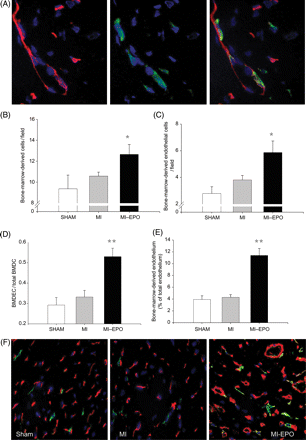
(A and B) Effect of erythropoietin treatment on incorporation of endothelial progenitor cells into the myocardial vasculature. (A) Bone-marrow-derived endothelial cells stained with hPAP (green), His 52 (endothelium, red), and DAPI (nucleus, blue) detected at 63% magnification. The three panels display the endothelium, bone-marrow-derived cells (green), and the fluorescent overlay of the same section, respectively, demonstrating endothelial cells that are bone-marrow-derived and appear yellow. (B) Bar graph representing the numbers of bone-marrow-derived cells (positive for hPAP) incorporated in the myocardium. (C) Bar graph representing the numbers of bone-marrow-derived endothelial cells (double positive for hPAP and His52) per high power field. (D) Bar graph representing the ratio between bone-marrow-derived endothelial cells to total bone-marrow-derived cells in the spared left ventricular free wall. (E) Bar graph representing the percentage of endothelium composed of bone-marrow-derived cells. (F) Representative fluorescent overlay of the treatment groups at 40 × magnification. Endothelial cells appear red, bone-marrow-derived cells appear green, bone-marrow-derived endothelial cells appear yellow. BMDC, bone-marrow-derived cells; BMDEC, bone-marrow-derived endothelial cells; hPAP, human placental alkaline phosphatase. *P < 0.05 vs. myocardial infarction, **P < 0.01 vs. myocardial infarction.
Erythropoietin induces vascular endothelial growth factor expression and upregulates its own receptor
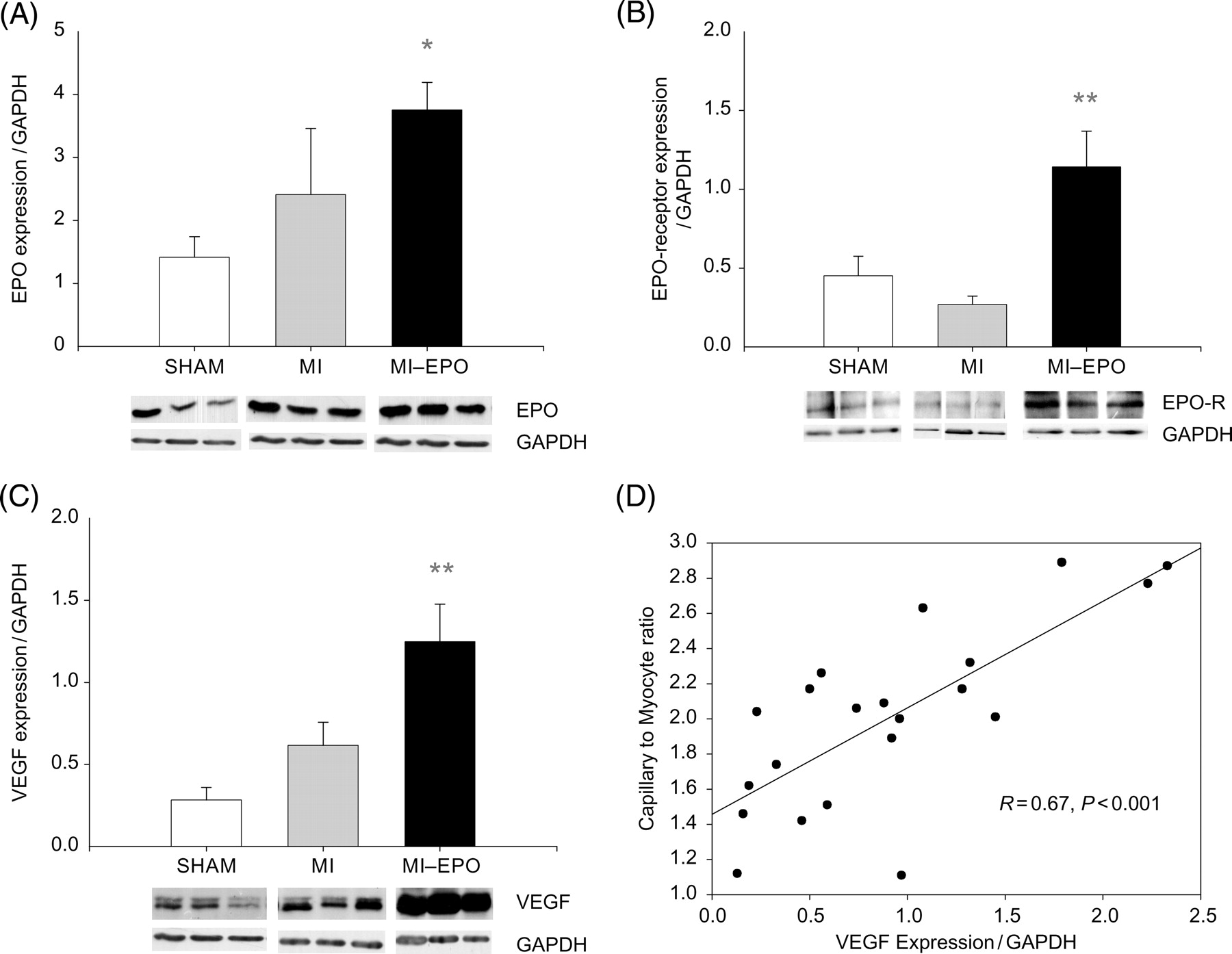
Three weeks after the last EPO administration, plasma EPO levels were comparable between MI groups [20 (2–76) vs. 18 (1–29) pg/mL in MI and MI–EPO groups, respectively), and slightly but non-significantly higher than sham [7 (1–30) pg/mL]. Myocardial expression of EPO was however two-fold higher in both MI groups (Figure 5, P < 0.05). EPO-receptor expression was slightly decreased in MI compared with sham group, EPO treatment resulted in a 3-fold upregulation of EPO-receptor expression (P < 0.01 vs. sham and MI, Figure 5).
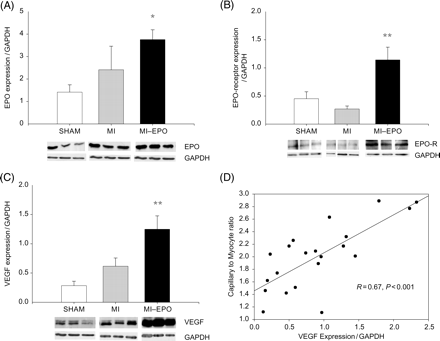
Expression of erythropoietin, erythropoietin-receptor, and vascular endothelial growth factor in the left ventricular free wall. (A) Bar graph and representative bands showing the difference in the expression of erythropoietin in the left ventricular free wall. (B) Bar graph and representative bands showing the difference in the expression of erythropoietin-receptor in the left ventricular free wall. (C) Bar graph and representative bands showing the difference in the expression of VEGF in the left ventricular free wall. (D) Scatter plot delineating the relation between capillary-to-myocyte ratio and VEGF expression in the left ventricular free wall of all groups (sham, myocardial infarction, and myocardial-infarction–erythropoietin). EPOR, erythropoietin-receptor; VEGF; vascular endothelial growth factor. *P < 0.05 vs. myocardial infarction, **P < 0.01 vs. myocardial infarction.
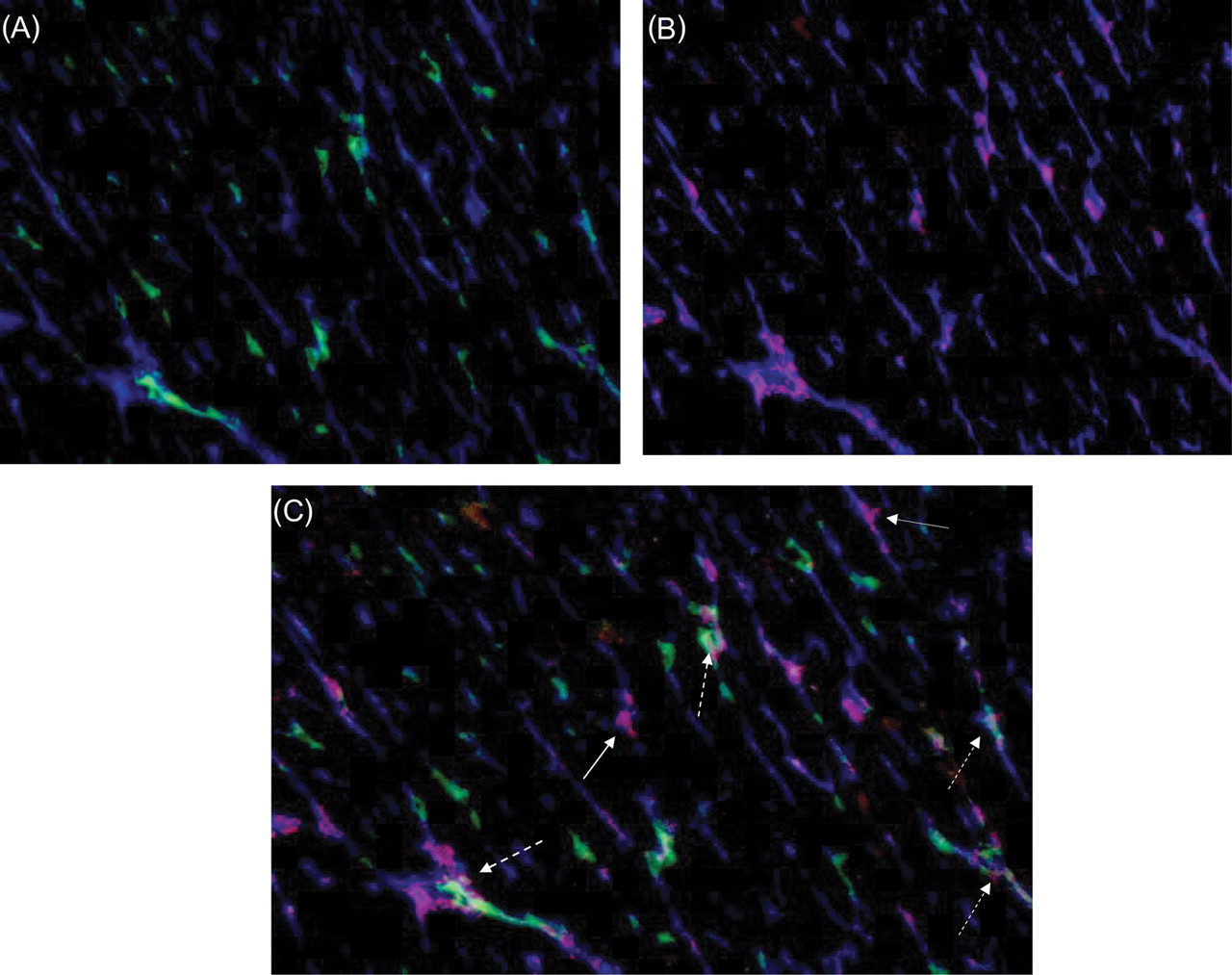
Although plasma VEGF levels were comparable among groups [7 (2–69), 12 (1–39), 8 (1–29) pg/mL in sham, MI, MI–EPO groups, respectively], a moderate increase in the expression of VEGF was observed in the MI-group (Figure 5, P = 0.07 trend), but expression of VEGF was increased 4.5-fold after EPO treatment (P < 0.01 vs. sham P < 0.02 vs. MI). The expression of VEGF was strongly correlated with new capillary growth (Figure 5, R = 0.67, P = 0.001). Triple staining with hPAP, His-52, and VEGF antibodies revealed that VEGF immunoreactivity was equally expressed in the endothelial cells of bone marrow and myocardial origin. (Figure 6).
![Expression of vascular endothelial growth factor by bone-marrow-derived and non-bone-marrow-derived endothelial cells. (A) Representative fluorescent overlay showing bone-marrow-derived endothelium [endothelium (his 52, blue) bone-marrow-derived cells (hPAP, green)]. (B) Representative fluorescent overlay showing vascular endothelial growth factor expression in the endothelium [endothelium (his 52, blue) vascular endothelial growth factor (red)]. (C) Merged picture of (A) and (B) showing that vascular endothelial growth factor is expressed by bone-marrow-derived (dashed arrow) and non-bone-marrow-derived (solid arrow) endothelial cells.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/28/16/10.1093/eurheartj/ehm177/2/m_ehm17706.gif?Expires=1716414840&Signature=Uzm-ygRKGCsTIQYDTNG6oS8vMs0zpmL71sNNXdE9aacfsalAeGgAOsCiwXFcQtLXrbWQDQYCB3cCq8rpbtZ3YK1Ehlpww7C8Or3DHWBB~~x0JYZH7dZOPvXHHFhN4Sbro1U-nmk8zb74k9bWWirO3RHzqpO8gFWSNk~~YSsAAF72MlRxaX-wLbhncKuiM1QW7c8hSFGM7Id32fRmFRxlvETyZMOuhwMsqG1UWUqsJJNHHifYclSOIW4YUOouH2nFhnqDZt9ErXNZHk2-20~xL913npKY2wrEwd2QqrW51gd6ji~YpsSwaqceRjYTX3-hXOzPEFq6sDVoInzMLMcmzA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of vascular endothelial growth factor by bone-marrow-derived and non-bone-marrow-derived endothelial cells. (A) Representative fluorescent overlay showing bone-marrow-derived endothelium [endothelium (his 52, blue) bone-marrow-derived cells (hPAP, green)]. (B) Representative fluorescent overlay showing vascular endothelial growth factor expression in the endothelium [endothelium (his 52, blue) vascular endothelial growth factor (red)]. (C) Merged picture of (A) and (B) showing that vascular endothelial growth factor is expressed by bone-marrow-derived (dashed arrow) and non-bone-marrow-derived (solid arrow) endothelial cells.
Discussion
The present study demonstrates for the first time that bone-marrow-derived progenitor cells are involved as a cardiac repair mechanism of systemic EPO treatment. EPO-induced neovascularization is associated with increased mobilization, myocardial homing, and vascular incorporation of EPC, which comprise 30% of the newly formed endothelium. The augmented EPC-mediated neovascularization resulted in a marked improvement of the myocardial microvascularization which was associated with attenuated MI-induced progression of LV-dilatation and decline in LV function. In addition, VEGF secretion in the myocardium was increased 4.5-fold by EPO, strongly correlating with neovascularization. VEGF was equally expressed by endothelial cells of myocardial and bone marrow origin, suggesting an additional auto-/paracrine mechanism distinct from bone-marrow-dependent neovascularization.
Erythropoietin prevents left-ventricular-dilatation and left-ventricular-dysfunction and regenerates the myocardial microvasculature, associated with endothelial progenitor cells mobilization
In the present study, we confirm that EPO improves cardiac function when administered in the chronic stage after MI. Similar to our previous study,11 EPO treatment starting 3 weeks after MI had no effect on myocardial infarct size or cardiomyocyte hypertrophy but restored capillary density to sham levels, suggesting that neovascularization is the predominant mechanism through which EPO improves the failing heart. Myocardial remodelling post-MI is associated with impaired perfusion of the non-infarcted LV wall due to disproportionate cardiomyocyte hypertrophy relative to (micro) vascular growth resulting in a progressive deterioration of cardiac function.24 Our data clearly demonstrate that administration of EPO restores the microvascularization and halts the deterioration of cardiac function. EPO-induced improvement of cardiac function, cardiac neovascularization, and EPC mobilization in post-MI LV-dysfunction was recently confirmed by two independent studies.9,12 Furthermore, in a distinct model of chronic myocardial dysfunction, EPO prevented doxorubicin induced cardiomyopathy, which was also linked to improved EPC mobilization and capillary density8,10 Thus, EPO consistently improves cardiac function in experimental models of CHF, strongly linked to increased EPC mobilization and improved microvascularization.
Endothelial progenitor cells mobilized by erythropoietin incorporate into newly formed capillaries
Our data are the first to demonstrate that EPC importantly contribute to EPO-mediated microvascular regeneration of the failing myocardium. The effects of EPO on EPC mobilization were first described by Heeschen et al.,18 who also demonstrated an association with neovascularization of ischaemic tissues. Recently, Urao et al.25 were the first to demonstrate that EPO increased incorporation of EPC into the endothelium, in a model of acute-aortic-wire injury. However, our data are the first to demonstrate that the EPC mobilized by EPO promote neovascularization of the hypertrophied myocardium in chronic post-MI LV dysfunction.
The importance of EPC in EPO-induced cardiac repair has been indirectly signified by three other reports. First, Prunier et al. showed that administration of EPO in a dose that does not induce EPC mobilization, failed to improve cardiac function and induce neovascularization, suggesting that EPC-mobilization is required for EPO to improve post-MI myocardial dysfunction.12 However, EPC were only characterized by expression of CD31, which is also expressed by a variety of leucocytes.26 Therefore, the data by Prunier et al. should be interpreted with caution. Secondly, in a model of doxorubicin-induced cardiotoxicity, infusion of isolated EPC prior to doxorubicin infusion, preserved cardiac function in a magnitude equal to EPO.10 Third, in a model of hypoxia-induced pulmonary hypertension, mice with erythroid restricted expression of the EPO-receptor demonstrated impaired EPC mobilization and augmented disease progression, which was restored by transplantation of wild-type bone marrow.27 Hence, EPC-mediated neovascularization appears to be crucial for EPO-induced protection from chronic vascular disease.
Erythropoietin increases vascular endothelial growth factor levels and induces expression of its own receptor
Thirty percent of the new endothelium was bone-marrow-derived indicating that the remaining new vessels originated from in situ proliferated endothelial cells. Both direct effects of EPO on in situ endothelial cell proliferation and paracrine functions of EPC may play a role in this process.28
In order to investigate whether EPO stimulated the paracrine effect of EPC, we evaluated the expression of the key angiogenic cytokine VEGF. We demonstrate that EPO treatment markedly increases expression of VEGF in the myocardium. Endothelial cells of myocardial and bone marrow origin demonstrated equal VEGF immunoreactivity, which indicates that EPO stimulates secretion of angiogenic factors by endothelial cells in an auto-/paracrine fashion irrespective of their origin. These findings suggest that in addition to EPC-mediated neovascularization, EPO increases in-situ proliferation of myocardial endothelial cells, providing a distinct parallel mechanisms of neovascularization. EPO-induced expression of VEGF has also been observed in cultured endothelial cells. Interestingly, neutralization of VEGF stopped the mitogenic effects of EPO in these cultures, suggesting that EPO-induced endothelial cell proliferation is dependent on VEGF.29,30 Moreover, in addition to auto-/paracrine effects on endothelial cells, myocardial upregulation of VEGF might have chemotactic effects on EPC. The increased VEGF expression would thereby stimulate homing of EPC into the myocardium, further augmenting EPC-mediated neovascularization. These data suggest an important role for VEGF in EPO-induced neovascularization. Further studies are needed to evaluate whether VEGF upregulation is a prerequisite for EPO-induced neovascularization. Although the exact role of VEGF remains to be established, our data are the first to demonstrate the broad range of pro-angiogenic effects exerted by EPO, namely EPC-mediated neovascularization, local upregulation of another key angiogenic factor (VEGF), and stimulation of in-situ proliferation of endothelial cells.
Furthermore, EPO increased the expression of its own receptor, possibly leading to increased sensitivity to EPO. This has previously been demonstrated in cultured endothelial cells where hypoxia and EPO synergistically induced EPO-receptor expression.31 These data suggest that EPO regulates EPO-receptor expression through a positive feedback mechanism. Basal expression-levels of EPO-receptor are relatively low in the non-erythropoietic tissues, but a marked up-regulation of the EPO-receptor has been demonstrated by acute metabolic stress, including hypoxia.32 Therefore, the elevated exogenous EPO levels during treatment might mimic a hypoxia related signal to increase EPO-receptor expression, further potentiating the effects of therapy.
Clinical implications
Our present and previous data clearly show that EPO-treatment prevents LV dilatation and myocardial functional decline by regenerating the myocardial microvasculature. Treatment with EPO has been identified as a promising and safe therapy for acute MI.33–35 Moreover, correction of anaemia in CHF with EPO has proved both safe and feasible.36 Our findings indicate, however, that EPO exerts beneficial haematocrit-independent effects, which might also prove beneficial for non-anaemic CHF patients. Nevertheless, unrestricted dosing regimens of EPO could elevate haematocrit to unacceptable levels, which might lead to impaired rheology, thrombogenicity and hypertension.37 Indeed, a significant increase in diastolic blood pressure was observed in our study, suggesting EPO-induced hypertension. However, increased arterial pressure might result from the improved myocardial contractility. Systolic and diastolic blood pressure were comparable between the MI–EPO and the sham group, supporting the hypothesis that the increased blood pressure is related to improved function. These limitations might be overcome by using dosing regimens that do not alter haematocrit levels.38 Alternatively, EPO-derivatives have recently emerged that do not ligate the EPO receptor on erythropoietic cells, but still confer tissue protection.39 Finally, a very recent study by Schneider et al.40 revealed that endocardial EPO injections improves the contractile function of hibernating myocardium, without affecting haematocrit levels.
Conclusion
EPO-induced myocardial neovascularization is mediated through a combination of EPC recruitment from the bone marrow and increased VEGF expression by endothelial cells.
Acknowledgements
We thank Azuwerus van Buiten, Maaike Goris, Liza Wong, Alex Kluppel, Bianca Meijeringh, Lennaert Kleijn, Martine Broekema, and Richard van Veghel for expert technical assistance and advice. B.D.W., van der Meer, and van der Harst are supported by ZonMW-NWO. E.L. and H.O. are supported by GUIDE. A.A.V. is a clinical established investigator of the Netherlands heart foundation Grant:2006T037.
Conflict of interest: none declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}