





Abstract
Aims To characterize the clinical profile of patients belonging to families affected with autosomal dominant arrhythmogenic right ventricular cardiomyopathy (ARVC) due to mutations of the gene encoding for the cell-to-cell adhesion protein desmoplakin (DSP).
Methods and results Thirty-eight subjects belonging to four families showing different DSP mutations (three missense and one in the intron–exon splicing region) underwent clinical and genetic investigation, including annual 12-lead ECG, signal averaged ECG, 24 h Holter ECG, and two-dimensional echocardiography. Twenty-six family members (11 males and 15 females) were found to carry a DSP mutation. After a follow-up of 1–24 years, median 6, 14 (54%) fulfilled (mean age at diagnosis 33±15 years) and 12 (mean age 43±24 years at the last follow-up) did not fulfil the established diagnostic criteria of ARVC, although five of them had some cardiac abnormalities. Clinical presentations were palpitations in six, sudden death (SD) in three, syncope in one, and chest pain with increased myocardial enzymes in two. Abnormal 12-lead ECG findings were present in 15 cases (58%), ventricular arrhythmias in 12 (46%), and late potentials in 11 (42%). Fourteen (54%) had abnormal echocardiographic findings, with left ventricular involvement in seven of them. SD occurred in six subjects and in three it was the first symptom of the disease; moreover, one subject died due to heart failure. The annual disease-related death and SD/aborted SD were 0.028 and 0.023 patient/year, respectively.
Conclusion Familial ARVC caused by DSP mutations is characterized by a high occurrence of SD even as first clinical manifestation. Left ventricular involvement is not a rare feature of the disease, which frequently escapes clinical diagnosis by applying the currently available criteria. Genetic screening is mandatory for early identification of asymptomatic carriers and preventive strategies within a family with a genotyped index case.
See for the editorial comment on this article (doi:10.1093/eurheartj/ehi343)
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a frequent familial myocardial disease with genetic heterogeneity.1 It is quite likely that the pathologic hallmark of the disease, a non-ischaemic death of the myocytes with fibro-fatty replacement constituting the anatomic basis for re-entry ventricular arrhythmias at risk of sudden death (SD), represents the common phenotype of several gene defects.2–5
Since the identification of the first ARVC locus in 1994,6 five disease genes have been identified so far.7–12 A 2 bp deletion in the plakoglobin has been proved to cause a recessive form of ARVC associated with palmoplantar keratosis and woolly hair, i.e. Naxos disease.7 Mutations of ryanodine receptor type 2 gene and regulatory mutations in transforming growth factor β3 gene have been found in ARVD2 and ARVD1, respectively.8,12 Moreover, mutations in the desmosomal proteins desmoplakin (DSP, ARVD8) and plakophilin-2 (ARVD9) have been discovered, in the absence of skin and hair abnormalities.9,11 DSP is the most represented protein of the desmosomes, which are cell-to-cell junctions particularly abundant in the keratinocytes and cardiomyocytes.13,14 Remarkably, mutations in the DSP gene have already been shown to underlie some autosomal dominant and recessive skin disorders, either with or without cardiac involvement.10,15–19 The identification of DSP mutations and the availability of genetic screening in affected family members enable us to gain an insight into the clinical features of the familial ARVD8 variant of the disease.
Methods
Genetic study
Twenty-five index cases were screened for DSP mutations. Polymerase chain reaction (PCR) primers flanking each exon of the human DSP gene were available.9 Mutation screening was performed by denaturing high-performance liquid chromatography and direct sequencing. All amplimers showing abnormal elution profiles were sequenced using the BIG DYE dideoxy-terminator chemistry on an ABI 3730XL DNA sequencer.
A control group of healthy and unrelated subjects (200 alleles) from the Italian population was used to exclude that the detected mutations were DNA polymorphisms. They were selected as first 100 subjects who responded to the advertisement of healthy control population recruitment and met all necessary criteria, i.e. absence of both personal and family history of heart disease.
The mutation screening was performed in all living family members of index cases in which a DSP mutation was detected.
In order to sequence the aberrant transcript of patient carrying the c.423-1G>A mutation, mRNA was isolated from 2.5 mL whole blood of the patient, using PAXgene™ Blood RNA kit (Qiagen, Hilden, Germany) following the supplier protocol. The corresponding cDNA was obtained from an RT–PCR reaction using 2 µg total RNA. The following primers were used to amplify the fragment corresponding to exons 3, 4, and 5. Primer forward: ATCAGAGAGATGCGGCAGAT; primer reverse: GCTGTTAATGTGCTGCTCCA.
Clinical study
The clinical study protocol included basal 12-lead ECG, signal averaged ECG (SAECG), 24 h Holter ECG, and two-dimensional echocardiography.20 These investigations were performed every 12 months. The clinical diagnosis of ARVC was put forward based on the criteria proposed by the ESC-ISFC.21
ECG, SAECG, and arrhythmias
Standard ECG was recorded at rest (25 mm/s, 10 mm/mV). The difference between the widest QRS complex in leads V1–V2–V3 minus that in V6 was estimated (QRS dispersion), and also the presence of inverted T wave in the precordial leads. Recorded ventricular arrhythmias were divided into the following: (1) ventricular fibrillation (VF); (2) sustained ventricular tachycardia (VT) (when lasting more than 30 s); (3) non-sustained VT; and (4) frequent premature ventricular complexes (PVCs) (>30/h). SAECG was performed using a MAC 15 system (Marquette Inc., Milwaukee, WI, USA); time domain analysis was obtained using three different filters at 25, 40, and 80 Hz.20
Echocardiography
M Mode, two-dimensional and Doppler echocardiography were done with Hewlett Packard Sonos 5500 system with S4 probe. Right and left ventricular end-diastolic and end-systolic volumes (RVEDV, RVESV, LVEDV, LVESV), right and left ventricular ejection fraction (RVEF, LVEF), and segmental or diffuse (≥two regions) kinetic abnormalities were evaluated. LVEDV and LVESV were calculated using an ellipsoid biplane area–length model derived from LV images in the apical four-chamber view.22 The ejection fraction was calculated using the following formula: end-diastolic volume minus end-systolic volume divided by end-diastolic volume. RVEDV and RVESV were calculated using an area–length method derived from orthogonal planes (apical four-chamber and subcostal long axis views).23 RVEDV was considered normal at 48±6 mL/m2.24 Structural progression was defined as a progressive alteration in ventricular dimensions and/or wall motion abnormalities during serial echocardiographic evaluation.25
Statistical analysis
Comparisons between patient groups were performed using a χ2 test or Fisher exact test for the categorical variables. All continuous variables were expressed as the mean value±1 SD for each measurement. All tests were two-tailed and a P-value <0.05 was considered statistically significant. Kaplan–Meier curves were produced to reveal the event-free survival rate during all life or up to ARVC diagnosis in DSP mutations carriers, considering all events (sustained VT, SD/aborted SD, heart failure). As patients were coming from only four families, the underlying assumption that all observations are independent could be violated. Therefore, we used nested design t-test and Kaplan-Meier curves considering family membership of each patient as a weighed variable. Statistical calculations were performed using Statistica version 6.0 (Statsoft, Tulsa, OK, USA).
Results
Identification of DSP mutations
DSP mutations have been identified in four out of 25 ARVC index cases (16%), randomly selected from a cohort of 100 probands. Then mutation screening was performed in all available family members, including first- and second-order relatives. Among 38 family members of the four index cases carrying DSP mutations, 26 (11 males and 15 females, mean age at the initial evaluation 35±20 years, minimum 8 to maximum 81 years) carried a DSP gene mutation (Figure 1 and Table 1).
Three novel mutations (R1775I, R1255K, and the splicing mutation c.423-1G>A) were identified, in addition to the previously reported one (S299R).9 None of the novel nucleotide changes were detected in 100 control subjects (200 chromosomes) from the same population. Mutations R1775I and R1255K are located in the rod domain, which is predicted to form the coiled-coil. The aminoacid changes occurred in residues which are conserved among species. The arginine/lysine aminoacid exchange could be considered significant, because the guanido group in the side-chain of arginine might be able to establish a greater number of hydrogen bonds with other aminoacids than the aminic group of lysine.
Mutation R1775I is located in the rod-domain of DSP, which is predicted to form the dimeric coil. Arginine is a hydrophilic aminoacid, whereas isoleucine is hydrophobic. This aminoacid change could destabilize the rod-domain preventing the formation of the coiled-coil. This view was confirmed by simulations obtained using PSIPRED.26
Mutation c.423-1G>A alters the acceptor splicing site of intron 3. Sequencing of the DSP transcript obtained from lymphocytes RNA of the proband showed the skipping of exon 4. This aberrant spliced mRNA contains a premature stop codon and should code for a truncated protein of 200 aminoacids in length.
In vitro studies are needed to evaluate the cell–cell adhesion effects of these mutant DSPs.
Main clinical and genetic data of DSP mutation carriers are summarized in Table 1. Among the 26 subjects carrying a DSP mutation, 14 (54%) (six males and eight females, age at diagnosis 33±15 years, minimum 15 to maximum 59 years) fulfilled and 12 (five males and seven females, mean age 43±24 years at the last follow-up) did not fulfil the established diagnostic criteria for ARVC. The distribution of major and minor criteria is reported in Table 1. Owing to the small size of the remaining three families, the penetrance was calculated only in family 105 (in the former two generations) as 64%.
Clinical presentation
Palpitations occurred in six patients, cardiac arrest in three, and syncope in one as the first symptom of the disease. Moreover, chest pain associated with ST segment elevation on basal ECG (Figure 2) and myocardial enzyme release, in the setting of angiographically normal coronary arteries, occurred in two siblings. No skin and hair abnormalities were found at physical examination.
ECG/SAECG findings
Major ECG and SAECG findings are summarized in Table 2. Abnormal ECG findings were present in 15 patients (58%). Negative T waves in the precordial leads were recorded in 12 (46%), QRS complex prolongation (>110 ms) in V1, V2, or V3 in seven (27%), incomplete right bundle branch block in five (19%), qS aspect in inferior leads in seven (27%), low QRS voltages in the peripheral leads in five (19%), and ST segment elevation (<2 mm) in four (15%) patients. QRS dispersion more than 20 ms was present in nine (35%) and epsilon waves in two subjects.
Late potentials were detected in 11 DSP mutation carriers (42%) and were evidenced at 25–40–80 Hz filters in six subjects, at 40–80 Hz in three patients, and at 80 Hz in two.
Ventricular arrhythmias
They were recorded in 12 subjects (46%) and ranged from VF (n=3) to monomorphic sustained VT with left bundle branch block morphology (n=4), non-sustained VT (n=2), and isolated monomorphic PVCs (n=3). In all subjects ventricular arrhythmias had left bundle branch block morphology, with variable QRS axis deviation. The total number of isolated PVCs ranged from 1000 to 3500/24 h. In two subjects (Patient 105, III, 10 and Patient 105, II, 4) sporadic isolated PVCs with right bundle branch block morphology were recorded.
Echocardiographic findings
Abnormal echocardiographic findings were present in 14 (54%) of DSP mutations carriers. An RV involvement was detected in 13 cases, showing a mean RVEDV of 81±30 mL/m2, a mean RVEF of 52±11%, and kinetic abnormalities involving one region in six and ≥2 regions in seven. An LV involvement was present in seven patients (mean age 35±19 years vs. 37±12 of those without LV involvement, P=0.830), which showed a mean LVEDV of 73±9 mL/m2, a mean LVEF of 53±8%, a mean RVEDV of 89±33 mL/m2 and a mean RVEF of 49±13%; LV kinetic abnormalities were diffuse in three and localized in four patients.
Considering only those subjects who fulfil the currently available diagnostic criteria (n=14), right ventricular abnormalities were detected in 86%, LV abnormalities in 43%, ventricular arrhythmias in 79%, SD/aborted SD in 36%, and heart failure in 14%.
DSP mutations carriers not fulfilling the ARVC diagnostic criteria
Twelve subjects did not fulfil the ESC-ISFC diagnostic criteria of ARVC. However, besides family history, five of them (42%) showed some cardiac signs, such as PVCs originating from the outflow tract of the RV (Patient 105, III, 7), LV kinetic alterations and positive late potentials (Patient 146, II, 2), regional RV abnormalities (Patient 151, III, 4) and positive late potentials (Patient 151, IV, 1), and qS in inferior leads (Patient 105, II, 1). This last patient died suddenly during follow-up. Noteworthy, no gender difference was found between DSP mutations carriers who fulfilled the ARVC diagnostic criteria (six out of 11 males, 55% vs. eight out of 15 females, 53%, P=0.677). Moreover, 33% of female DSP carriers did not present any cardiac sign vs. 18% of male (P=0.07).
Therapy
Eight patients underwent antiarrhythmic therapy, i.e. amiodarone (either alone or in association with beta-blocker), dysopiramide in association with beta-blocker, propafenone, and sotalol; two received an implantable cardioverter defibrillator (ICD).
Follow-up
During follow-up (range 1–24 years, median 6 years), six mutation carriers who had been considered normal at the first evaluation (mean age 23±10 years), developed symptoms of the disease (mean age at diagnosis 28±10). A structural progression on echocardiography was detected in 23%, and on ECG and/or SAECG in 31% of DSP carriers. During this period, three patients had SD, two had aborted SD, and one died due to heart failure. The annual disease-related and SD/aborted SD mortality were 0.028 and 0.023 patient/year, respectively. Kaplan–Meier curves for event-free survival rate in DSP mutations carriers are indicated in Figure 3. By the age of 45, the event-free survival was 58%.
SD/aborted SD
SD was found in six subjects, occurring at rest in all but one case, and in three it was the first symptom of the disease. Patient 105, II, 1 died suddenly at the age of 65, without ECG criteria for ARVC. Patient 105, III, 11 experienced an aborted SD at the age of 18. He was first admitted to the hospital at the age of 12, because of prolonged chest pain with ST segment elevation and negative T waves in leads V1–V3 and increased myocardial enzymes [peak serum creatine phosphokinase (CPK): 415 U/L]. Angiography demonstrated RV posterior wall and apical kinetic abnormalities and mild hypokinesia of the posterior LV wall with normal volumes and patent coronary arteries.
Patient 105, IV, 4, who had been considered unaffected during familial screening at the age of 13, died suddenly 2 years later. The retrospective analysis of a 12-lead resting ECG, performed 8 months before death for sports eligibility, showed incomplete right bundle branch block with mild ST segment elevation in V1–V2, negative T wave in V1, and Q wave in D2, D3, and AVF (Figure 4).
Patient 146, III, 1 died at the age of 33 after recurrent left bundle branch block morphology with left axis deviation sustained VT, which was haemodynamically well tolerated, while waiting for ICD implantation on amiodarone and beta-blocker therapy. Twelve-lead ECG performed 6 months before death showed negative T wave in V4–V6, RV conduction delay, and low QRS voltages (Figure 5).
Patient 151, III, 2, who had been diagnosed at the age of 37 due to ECG alterations, had a syncopal episode with loss of consciousness 5 years later, on sotalol therapy, and a sustained VT was recorded. After this episode an ICD was implanted that discharged appropriately several times (sustained VT, 190 bpm). The patient is now also on disopyramide and beta-blocker therapy.
Finally, Patient 152, II, 1 had suffered VF on effort as first symptom at the age of 40 years and was successfully resuscitated. Following the event, an ICD was implanted.
Autopsy findings
The heart was available for a detailed postmortem examination in two patients. In Patient 105, IV, 4 the heart weight was 300 g, and no evidence of coronary artery and valve disease, myocardial hypertrophy, cavity enlargement, and aneurysm formation was found. An ‘infarct-like’ band of acute–subacute myocyte necrosis associated with inflammation was evident in the outer mid–subepicardial layer of the postero-septal and postero-lateral walls of the LV (Figure 4B and C). The inflammatory infiltrate was polymorphous and was associated with contraction band necrosis, myocytolysis, granulation tissue, and loose fibrous and fatty tissue repair. Foci of coagulative and contraction band necrosis were also present in the RV myocardium with spotty intramural fatty infiltration. In Patient 146, III, 1 the heart weight was increased (500 g) and no evidence of coronary artery and valve disease was found. RV dilatation together with inferior and infundibular aneurysms was present. At histology, an extensive myocardial atrophy with transmural fibro-fatty tissue replacement was visible at the level of antero-lateral, inferior, and infundibular walls of the RV, as well as in the mid-outer thirds of the anterior, lateral, antero-septal, and postero-septal walls of the LV (Figure 5). Myocytolysis and lymphocytic inflammatory infiltrates were also detected.
Discussion
The clinical features of autosomal dominant familial ARVC due to four different DSP mutations are herein reported. The clinical spectrum is quite heterogeneous and, although some ECG and/or echocardiographic abnormalities were present in two-thirds of gene mutation carriers, nearly half of them did not fulfil the established diagnostic criteria for ARVC. Half of the patients with echocardiographic abnormalities also showed LV involvement. However, on the basis of the family history of ARVC, ECG features (negative T waves in the precordial leads, RV conduction delay, epsilon wave, low QRS voltages in peripheral leads), presence of late potentials at SAECG, ventricular arrhythmias with left bundle branch block morphology, and echocardiographic features with predominant RV dilatation, as well as more depressed RV ejection fraction when compared with the left one, together with RV segmental abnormalities, a typical form of dilated cardiomyopathy could be excluded. In our series, the presence of ARVC based on the diagnostic criteria was higher in male than in female DSP mutations carriers, confirming previous data from both clinical and autopsy series.3–5,11,20 This could be interpreted either as a gender-related penetrance or as the consequence of additional acquired and genetic factors involved in disease expression.
The annual disease-related and SD mortality were 0.028 and 0.023 patient/year, respectively, which is similar to that reported in Naxos disease.25
ARVC and intercellular junction protein mutations
DSP is a protein of the desmosomes, which are intercellular junctions particularly abundant in the epidermidis and heart muscle, where they play an important role for cell rigidity and strength.26–29 A deletion of another cell adhesion protein, i.e. plakoglobin, has been identified in Naxos disease, a recessive syndrome in which ARVC is associated with palmoplantar keratosis and woolly hair.7,25,30 More recently, mutations of plakophilin-2 have been found in 27% of unrelated ARVC probands.11 Mutations of DSP gene have already been shown to underlie autosomal diseases characterized by hair and skin disorders, either alone or in association with cardiac involvement.15–19 Although in the Carvajal syndrome a cardiac phenotype of dilated cardiomyopathy has been reported,16–18 a clinical picture of ARVC has been recently described in a Muslim-Arab family from Jerusalem.10
Different from these recessive cardio-cutaneous syndromes, ARVD8 has a dominant pattern of inheritance, in which heterozygous carriers of the mutation show typical cardiac abnormalities, in the absence of macroscopic skin and hair involvement. However, dominant DSP mutations associated only with skin abnormalities have also been described.15,19 The mutation Q331X reported by Armstrong et al.,15 as the one by Wittock et al.,19 resulted in haploinsufficiency and led to palmoplantar keratosis. The reason why mutation c.423-1G>A leads to ARVC, without macroscopic evidence of keratosis, is still unknown and functional studies are required.
In the original Carvajal paper, low voltage QRS complex were present in all but one patient, inverted T wave in the precordial and inferior leads in half, and ventricular arrhythmias in all,17 clinical features more in keeping with ARVC than with dilated cardiomyopathy. Moreover, the pathology of the heart in Carvajal syndrome disclosed RV aneurysms in the ‘triangle of dysplasia’ as well as massive LV involvement, all features in keeping with biventricular ARVC, although in the absence of substantial fatty infiltration.31
Validation of ARVC diagnostic criteria
Although our study is not dealing with unrelated ARVC index patients carrying different DSP mutations, which might result in a broader spectrum of clinical features, our patient selection gives the opportunity to evaluate the entire clinical spectrum of the disease from concealed forms to the overt ones. Nearly half of DSP mutations carriers did not fulfil the established diagnostic criteria for ARVC.21 However, 42% of them had a few cardiac signs, not diagnostic for ARVC, and one of them died suddenly during the follow-up showing only q waves in inferior leads. Noteworthy, a q wave in inferior leads was present in more than one-fourth of DSP carriers and it could be related to the presence of fibrosis in the inferior LV wall, as evidenced by clinical–pathologic correlation in Patient 105, IV, 4. These findings allow us to speculate whether the established criteria are too restrictive in the setting of a familial disease where unspecific instrumental abnormalities could have otherwise a high probability of being the expression of a gene defect. By applying the broadened criteria proposed by Hamid et al.,32 four additional family members might have been considered affected. Our study confirms that genetic screening for DSP mutation is a valuable tool for early pre-symptomatic diagnosis of ARVC within a family with a genotyped index case, as many affected family members were diagnosed during family screening and remained fully asymptomatic. However, we must admit that, with the advent of genetic testing, a growing number of asymptomatic carriers with mild or subclinical disease expression will be identified with obvious management dilemmas. We suggest the following plan for asymptomatic gene carriers: six monthly follow-up, avoidance of strenuous physical activity, and advice of reporting any symptom to a cardiologist. Moreover, we must be aware that this strategy would probably not have prevented some of the adverse events documented in our population, given that SD may occur at rest, without prominent clinical findings, and in the absence of warning symptoms.
Histopathologic findings
The features are quite unique as autopsy was performed in two cases, with and without a clinical diagnosis of ARVC, respectively. In the former, the autopsy findings, consisting of transmural fibro-fatty replacement with aneurysms formation, were in keeping with a typical form of ARVC, whereas in the latter the arrhythmogenic substrates of the disease at early stage were documented, consisting of an extensive myocardial necrosis together with inflammation, loose fibrous, and fatty tissue repair. Noteworthy, in two additional patients the disease onset was characterized by an ‘infarct-like’ picture with chest pain, ST segment ECG changes, and myocardial enzyme release. These data further support the hypothesis that these symptoms/signs could be a manifestation of a myocyte necrosis occurring as a part of the disease process. To this regard, we previously demonstrated in ARVC that myocardial destruction with replacement by fat may be episodic rather than gradual and continuous and these bursts are characterized by acute symptoms and signs.33
The absence of either wall thinning or aneurysms, which are considered the morphologic hallmarks of ARVC, is explained by the recent onset with early subepicardial location of the myocyte injury and repair process, in the absence of transmural extension towards the endocardium. These results broaden our concept of the histologic spectrum of ARVC. As DSP is present in all cardiomyocytes, there is no reason for a preferential RV involvement. Noteworthy, both heart specimens had extensive LV involvement. Owing to the thinner wall, the RV involvement may manifest clinically earlier than the LV one in terms of kinetic abnormalities and cavity enlargement, although the pathologic process is already going on in both ventricles. Another potential mechanism of preferential RV manifestation of ARVC might be related to the higher propensity to stretch-related rupture of vulnerable cell–cell contact due to latent damage caused by heterozygous mutations in genes encoding desmosomal proteins.
Conclusions
Familial ARVC due to DSP mutations is characterized by a high occurrence of SD even in the concealed phase and LV involvement is not a rare feature. The disease frequently escapes clinical recognition by strictly using the established diagnostic criteria. Genetic screening may be of help for early identification of asymptomatic carriers and preventive strategies within a family with a genotyped index case.
Acknowledgements
This study was supported by Ministry of Health and MURST, Rome; Fondazione Cassa di Risparmio, Padova e Rovigo; ARVC/D Project, QLG1-CT-2000-01091 Fifth Framework Programme European Commission, Bruxelles. B.B. is a recipient of a research temporary position pursuant to ARVC/D Project, European Commission. The authors are deeply indebted to Mrs Paola Marcon for her help in collecting familial data and to Mikolaj Winnicki, MD for statistical analysis support.
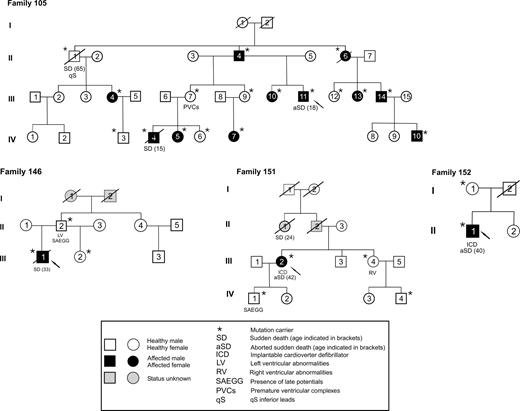
Figure 1 Family pedigrees of the four ARVC index cases carrying a DSP mutation.
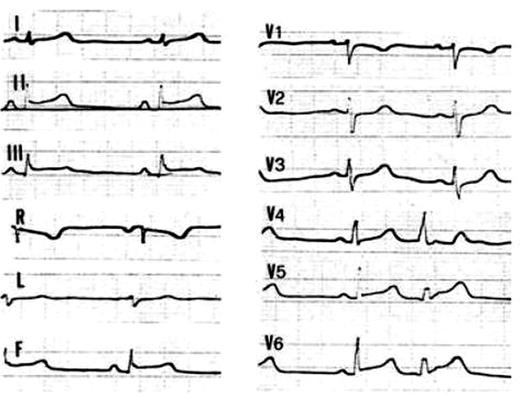
Figure 2 Basal 12-lead ECG tracing in Patient 105, III, 10 who presented with chest pain associated with increased serum CPK markers at the age of 17: note the ST segment elevation in the inferior and left lateral leads.
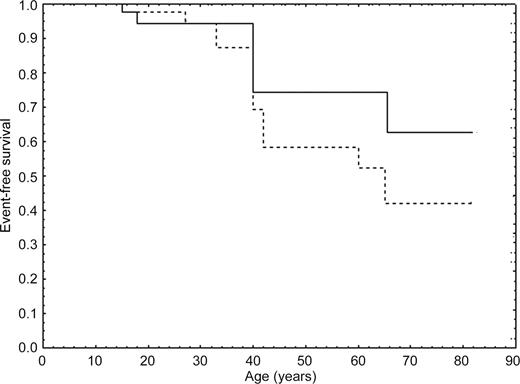
Figure 3 Event-free survival curve in all subjects carrying DSP mutations for all events (sustained VT, SD/aborted SD, HF) using nested design Kaplan–Meier, considering either their all life (dotted lines) or time up to ARVC diagnosis as the end of the observation (continuous line).
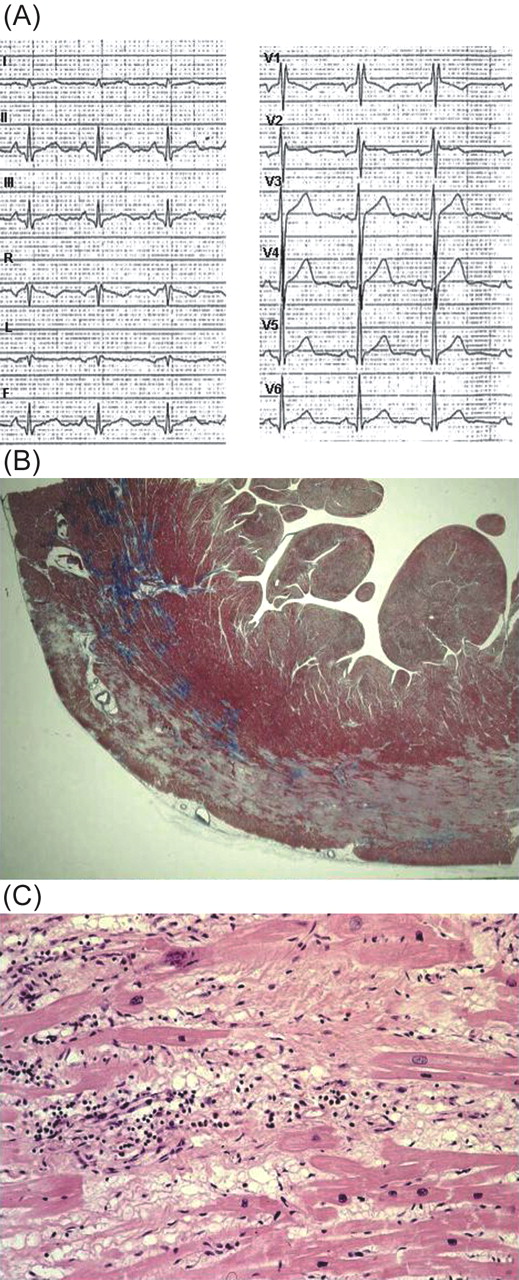
Figure 4 Patient 105, IV, 4, a 15-year-old asymptomatic boy carrying DSP mutation who died suddenly at rest. (A) 12-lead ECG tracing recorded 8 months before death at pre-participation screening for sports eligibility: note the presence of incomplete right bundle branch block and a +70° QRS axis. Mild ST segment elevation in V1–V2, inverted T wave in V1, and Q wave in the inferior leads are also present. (B) Panoramic histologic view of the postero-lateral left ventricular wall showing a subepicardial band of acute–subacute myocyte necrosis with loose fibrous tissue and granulation tissue (Trichrome Heidenhain ×3). (C) Myocyte necrosis, myocytolysis, and polymorphous inflammatory infiltrates together with fibrous and fatty tissue repair are visible at higher magnification (haematoxylin–eosin ×40).
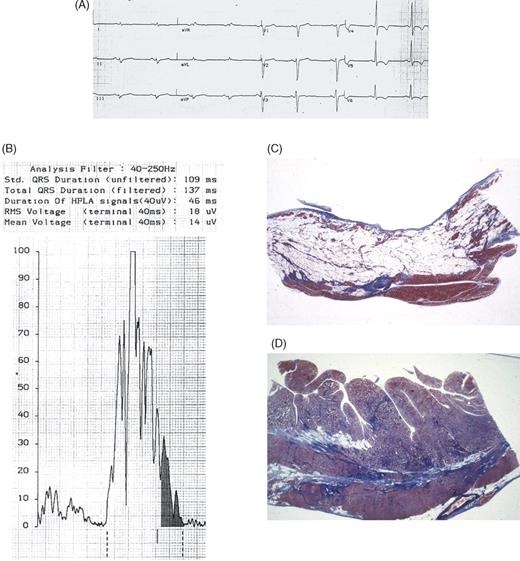
Figure 5 Patient 146, III, 1, a 33-year-old man with an overt form of ARVC who died suddenly while waiting for ICD implantation despite antiarrhythmic therapy. (A) 12-lead ECG showing negative T wave in V4–V6, right ventricular conduction delay and low QRS voltages. (B) SAECG: note the presence of late potentials at 40/250 Hz filter. (C) Panoramic histologic section of the anterior right ventricular wall: extensive transmural fibro-fatty replacement with myocardial atrophy is evident (Trichrome Heidenhain ×3). (D) Panoramic histologic section of the lateral left ventricular wall: fibro-fatty replacement with prevalent fibrous tissue is also evident (Trichrome Heidenhain ×3).
Clinical findings of 26 subjects carrying DSP mutations
| Case | DSP mutations | Age at diagnosis | Sex | Initial evaluation | Age at first symptom (years) | Follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | AA change | Age (years) | QRD (ms) | T–wave inversion | Clinical events | Therapy | Structural progression | Outcome | Duration (years) | Criteria Mj/m | ||||
| 105, II, 1 | c.897C>G | S299R | M | 63 | 0 | — | 65 (SD) | SD | — | — | SD | 2 | 1/0 | |
| 105, II, 4 | c.897C>G | S299R | 53 | M | 53 | 80 | V1–V5 | 20 (syncope) | HF | Amiodarone+BB | RV, LV | HF | 23 | 3/2 |
| 105, II, 6 | c.897C>G | S299R | 55 | F | 55 | 80 | V1–V5 | 55 (palpit.) | HF | Amiodarone | RV, LV | Death of HF | 24 | 3/2 |
| 105, III, 4 | c.897C>G | S299R | 34 | F | 32 | 20 | V1–V3 | — | — | — | — | Asymptomatic | 7 | 1/3 |
| 105, III, 7 | c.897C>G | S299R | F | 47 | 40 | — | 48 (palpit.) | — | — | — | Asymptomatic | 6 | 1/1 | |
| 105, III, 9 | c.897C>G | S299R | F | 48 | 40 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, III,10 | c.897C>G | S299R | 17 | F | 12 | 40 | V1–V2 | 17 (chest pain) | — | Propafenone | — | Asymptomatic | 15 | 1/4 |
| 105, III,11 | c.897C>G | S299R | 18 | M | 12 | 40 | V1–V5 | 12 (chest pain) | VF, aSD | Disop.+BB | RV, LV | Asymptomatic | 22 | 3/2 |
| 105, III,12 | c.897C>G | S299R | F | 51 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | |
| 105, III,13 | c.897C>G | S299R | 59 | F | 59 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/3 |
| 105, III,14 | c.897C>G | S299R | 44 | M | 40 | 20 | — | — | — | — | — | Asymptomatic | 19 | 1/4 |
| 105, IV, 3 | c.897C>G | S299R | M | 8 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/0 | |
| 105, IV, 4 | c.897C>G | S299R | 15 | M | 13 | 20 | — | 15 (VF,SD) | VF, SD | — | — | SD (autopsy) | 2 | 1/2 |
| 105, IV, 5 | c.897C>G | S299R | 27 | F | 21 | 20 | V1–V3 | 21 (palpit.) | — | — | — | Asymptomatic | 6 | 1/3 |
| 105, IV, 6 | c.897C>G | S299R | F | 14 | 20 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, IV, 7 | c.897C>G | S299R | 21 | F | 21 | 20 | — | 21 (palpit.) | NSVT | Sotalol | RV | Asymptomatic | 6 | 2/2 |
| 105, IV, 10 | c.897C>G | S299R | 21 | F | 21 | 20 | — | — | — | — | — | Asymptomatic | 10 | 2/1 |
| 146, II, 2 | c.423-1G>A | M | 58 | 30 | — | — | — | — | — | Asymptomatic | 6 | 1/1 | ||
| 146, III, 1 | c.423-1G>A | 25 | M | 25 | 40 | V4–V6 | 25 (palpit.) | VT, SD | Amiodarone+BB | RV | SD (autopsy) | 8 | 2/2 | |
| 146, III, 2 | c.423-1G>A | F | 10 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | ||
| 151, III, 2 | c.5324G>T | R1775I | 37 | F | 37 | 30 | V1–V4 | 38 (palpit.) | VT, aSD | ICD, Disop.+BB | RV | Asymptomatic | 5 | 1/3 |
| 151, III, 4 | c.5324G>T | R1775I | F | 52 | 20 | — | — | — | — | — | Asymptomatic | 1 | 0/2 | |
| 151, IV, 1 | c.5324G>T | R1775I | M | 13 | 0 | — | — | — | — | — | Asymptomatic | 3 | 0/2 | |
| 151, IV, 4 | c.5324G>T | R1775I | M | 18 | 0 | V1 | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, I, 1 | c.3764G>A | R1255K | F | 81 | 0 | — | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, II, 1 | c.3764G>A | R1255K | 40 | M | 40 | 0 | V1 | 40 (aSD) | — | ICD, sotalol | — | Asymptomatic | 2 | 1/3 |
| Case | DSP mutations | Age at diagnosis | Sex | Initial evaluation | Age at first symptom (years) | Follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | AA change | Age (years) | QRD (ms) | T–wave inversion | Clinical events | Therapy | Structural progression | Outcome | Duration (years) | Criteria Mj/m | ||||
| 105, II, 1 | c.897C>G | S299R | M | 63 | 0 | — | 65 (SD) | SD | — | — | SD | 2 | 1/0 | |
| 105, II, 4 | c.897C>G | S299R | 53 | M | 53 | 80 | V1–V5 | 20 (syncope) | HF | Amiodarone+BB | RV, LV | HF | 23 | 3/2 |
| 105, II, 6 | c.897C>G | S299R | 55 | F | 55 | 80 | V1–V5 | 55 (palpit.) | HF | Amiodarone | RV, LV | Death of HF | 24 | 3/2 |
| 105, III, 4 | c.897C>G | S299R | 34 | F | 32 | 20 | V1–V3 | — | — | — | — | Asymptomatic | 7 | 1/3 |
| 105, III, 7 | c.897C>G | S299R | F | 47 | 40 | — | 48 (palpit.) | — | — | — | Asymptomatic | 6 | 1/1 | |
| 105, III, 9 | c.897C>G | S299R | F | 48 | 40 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, III,10 | c.897C>G | S299R | 17 | F | 12 | 40 | V1–V2 | 17 (chest pain) | — | Propafenone | — | Asymptomatic | 15 | 1/4 |
| 105, III,11 | c.897C>G | S299R | 18 | M | 12 | 40 | V1–V5 | 12 (chest pain) | VF, aSD | Disop.+BB | RV, LV | Asymptomatic | 22 | 3/2 |
| 105, III,12 | c.897C>G | S299R | F | 51 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | |
| 105, III,13 | c.897C>G | S299R | 59 | F | 59 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/3 |
| 105, III,14 | c.897C>G | S299R | 44 | M | 40 | 20 | — | — | — | — | — | Asymptomatic | 19 | 1/4 |
| 105, IV, 3 | c.897C>G | S299R | M | 8 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/0 | |
| 105, IV, 4 | c.897C>G | S299R | 15 | M | 13 | 20 | — | 15 (VF,SD) | VF, SD | — | — | SD (autopsy) | 2 | 1/2 |
| 105, IV, 5 | c.897C>G | S299R | 27 | F | 21 | 20 | V1–V3 | 21 (palpit.) | — | — | — | Asymptomatic | 6 | 1/3 |
| 105, IV, 6 | c.897C>G | S299R | F | 14 | 20 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, IV, 7 | c.897C>G | S299R | 21 | F | 21 | 20 | — | 21 (palpit.) | NSVT | Sotalol | RV | Asymptomatic | 6 | 2/2 |
| 105, IV, 10 | c.897C>G | S299R | 21 | F | 21 | 20 | — | — | — | — | — | Asymptomatic | 10 | 2/1 |
| 146, II, 2 | c.423-1G>A | M | 58 | 30 | — | — | — | — | — | Asymptomatic | 6 | 1/1 | ||
| 146, III, 1 | c.423-1G>A | 25 | M | 25 | 40 | V4–V6 | 25 (palpit.) | VT, SD | Amiodarone+BB | RV | SD (autopsy) | 8 | 2/2 | |
| 146, III, 2 | c.423-1G>A | F | 10 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | ||
| 151, III, 2 | c.5324G>T | R1775I | 37 | F | 37 | 30 | V1–V4 | 38 (palpit.) | VT, aSD | ICD, Disop.+BB | RV | Asymptomatic | 5 | 1/3 |
| 151, III, 4 | c.5324G>T | R1775I | F | 52 | 20 | — | — | — | — | — | Asymptomatic | 1 | 0/2 | |
| 151, IV, 1 | c.5324G>T | R1775I | M | 13 | 0 | — | — | — | — | — | Asymptomatic | 3 | 0/2 | |
| 151, IV, 4 | c.5324G>T | R1775I | M | 18 | 0 | V1 | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, I, 1 | c.3764G>A | R1255K | F | 81 | 0 | — | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, II, 1 | c.3764G>A | R1255K | 40 | M | 40 | 0 | V1 | 40 (aSD) | — | ICD, sotalol | — | Asymptomatic | 2 | 1/3 |
AA, aminoacidic change; aSD, aborted SD; BB, beta-blockers; Disop., disopyramide; HF, heart failure; ICD, implantable cardioverter defibrillator; LV, left ventricle; Mj, major; m, minor; NSVT, non sustained ventricular tachycardia; palpit., palpitations; QRD, QRS dispersion; RV, right ventricle; SD, sudden death; VF, ventricular fibrillation; VT, sustained ventricular tachycardia.
Clinical findings of 26 subjects carrying DSP mutations
| Case | DSP mutations | Age at diagnosis | Sex | Initial evaluation | Age at first symptom (years) | Follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | AA change | Age (years) | QRD (ms) | T–wave inversion | Clinical events | Therapy | Structural progression | Outcome | Duration (years) | Criteria Mj/m | ||||
| 105, II, 1 | c.897C>G | S299R | M | 63 | 0 | — | 65 (SD) | SD | — | — | SD | 2 | 1/0 | |
| 105, II, 4 | c.897C>G | S299R | 53 | M | 53 | 80 | V1–V5 | 20 (syncope) | HF | Amiodarone+BB | RV, LV | HF | 23 | 3/2 |
| 105, II, 6 | c.897C>G | S299R | 55 | F | 55 | 80 | V1–V5 | 55 (palpit.) | HF | Amiodarone | RV, LV | Death of HF | 24 | 3/2 |
| 105, III, 4 | c.897C>G | S299R | 34 | F | 32 | 20 | V1–V3 | — | — | — | — | Asymptomatic | 7 | 1/3 |
| 105, III, 7 | c.897C>G | S299R | F | 47 | 40 | — | 48 (palpit.) | — | — | — | Asymptomatic | 6 | 1/1 | |
| 105, III, 9 | c.897C>G | S299R | F | 48 | 40 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, III,10 | c.897C>G | S299R | 17 | F | 12 | 40 | V1–V2 | 17 (chest pain) | — | Propafenone | — | Asymptomatic | 15 | 1/4 |
| 105, III,11 | c.897C>G | S299R | 18 | M | 12 | 40 | V1–V5 | 12 (chest pain) | VF, aSD | Disop.+BB | RV, LV | Asymptomatic | 22 | 3/2 |
| 105, III,12 | c.897C>G | S299R | F | 51 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | |
| 105, III,13 | c.897C>G | S299R | 59 | F | 59 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/3 |
| 105, III,14 | c.897C>G | S299R | 44 | M | 40 | 20 | — | — | — | — | — | Asymptomatic | 19 | 1/4 |
| 105, IV, 3 | c.897C>G | S299R | M | 8 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/0 | |
| 105, IV, 4 | c.897C>G | S299R | 15 | M | 13 | 20 | — | 15 (VF,SD) | VF, SD | — | — | SD (autopsy) | 2 | 1/2 |
| 105, IV, 5 | c.897C>G | S299R | 27 | F | 21 | 20 | V1–V3 | 21 (palpit.) | — | — | — | Asymptomatic | 6 | 1/3 |
| 105, IV, 6 | c.897C>G | S299R | F | 14 | 20 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, IV, 7 | c.897C>G | S299R | 21 | F | 21 | 20 | — | 21 (palpit.) | NSVT | Sotalol | RV | Asymptomatic | 6 | 2/2 |
| 105, IV, 10 | c.897C>G | S299R | 21 | F | 21 | 20 | — | — | — | — | — | Asymptomatic | 10 | 2/1 |
| 146, II, 2 | c.423-1G>A | M | 58 | 30 | — | — | — | — | — | Asymptomatic | 6 | 1/1 | ||
| 146, III, 1 | c.423-1G>A | 25 | M | 25 | 40 | V4–V6 | 25 (palpit.) | VT, SD | Amiodarone+BB | RV | SD (autopsy) | 8 | 2/2 | |
| 146, III, 2 | c.423-1G>A | F | 10 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | ||
| 151, III, 2 | c.5324G>T | R1775I | 37 | F | 37 | 30 | V1–V4 | 38 (palpit.) | VT, aSD | ICD, Disop.+BB | RV | Asymptomatic | 5 | 1/3 |
| 151, III, 4 | c.5324G>T | R1775I | F | 52 | 20 | — | — | — | — | — | Asymptomatic | 1 | 0/2 | |
| 151, IV, 1 | c.5324G>T | R1775I | M | 13 | 0 | — | — | — | — | — | Asymptomatic | 3 | 0/2 | |
| 151, IV, 4 | c.5324G>T | R1775I | M | 18 | 0 | V1 | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, I, 1 | c.3764G>A | R1255K | F | 81 | 0 | — | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, II, 1 | c.3764G>A | R1255K | 40 | M | 40 | 0 | V1 | 40 (aSD) | — | ICD, sotalol | — | Asymptomatic | 2 | 1/3 |
| Case | DSP mutations | Age at diagnosis | Sex | Initial evaluation | Age at first symptom (years) | Follow-up | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | AA change | Age (years) | QRD (ms) | T–wave inversion | Clinical events | Therapy | Structural progression | Outcome | Duration (years) | Criteria Mj/m | ||||
| 105, II, 1 | c.897C>G | S299R | M | 63 | 0 | — | 65 (SD) | SD | — | — | SD | 2 | 1/0 | |
| 105, II, 4 | c.897C>G | S299R | 53 | M | 53 | 80 | V1–V5 | 20 (syncope) | HF | Amiodarone+BB | RV, LV | HF | 23 | 3/2 |
| 105, II, 6 | c.897C>G | S299R | 55 | F | 55 | 80 | V1–V5 | 55 (palpit.) | HF | Amiodarone | RV, LV | Death of HF | 24 | 3/2 |
| 105, III, 4 | c.897C>G | S299R | 34 | F | 32 | 20 | V1–V3 | — | — | — | — | Asymptomatic | 7 | 1/3 |
| 105, III, 7 | c.897C>G | S299R | F | 47 | 40 | — | 48 (palpit.) | — | — | — | Asymptomatic | 6 | 1/1 | |
| 105, III, 9 | c.897C>G | S299R | F | 48 | 40 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, III,10 | c.897C>G | S299R | 17 | F | 12 | 40 | V1–V2 | 17 (chest pain) | — | Propafenone | — | Asymptomatic | 15 | 1/4 |
| 105, III,11 | c.897C>G | S299R | 18 | M | 12 | 40 | V1–V5 | 12 (chest pain) | VF, aSD | Disop.+BB | RV, LV | Asymptomatic | 22 | 3/2 |
| 105, III,12 | c.897C>G | S299R | F | 51 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | |
| 105, III,13 | c.897C>G | S299R | 59 | F | 59 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/3 |
| 105, III,14 | c.897C>G | S299R | 44 | M | 40 | 20 | — | — | — | — | — | Asymptomatic | 19 | 1/4 |
| 105, IV, 3 | c.897C>G | S299R | M | 8 | 0 | — | — | — | — | — | Asymptomatic | 1 | 1/0 | |
| 105, IV, 4 | c.897C>G | S299R | 15 | M | 13 | 20 | — | 15 (VF,SD) | VF, SD | — | — | SD (autopsy) | 2 | 1/2 |
| 105, IV, 5 | c.897C>G | S299R | 27 | F | 21 | 20 | V1–V3 | 21 (palpit.) | — | — | — | Asymptomatic | 6 | 1/3 |
| 105, IV, 6 | c.897C>G | S299R | F | 14 | 20 | — | — | — | — | — | Asymptomatic | 6 | 1/0 | |
| 105, IV, 7 | c.897C>G | S299R | 21 | F | 21 | 20 | — | 21 (palpit.) | NSVT | Sotalol | RV | Asymptomatic | 6 | 2/2 |
| 105, IV, 10 | c.897C>G | S299R | 21 | F | 21 | 20 | — | — | — | — | — | Asymptomatic | 10 | 2/1 |
| 146, II, 2 | c.423-1G>A | M | 58 | 30 | — | — | — | — | — | Asymptomatic | 6 | 1/1 | ||
| 146, III, 1 | c.423-1G>A | 25 | M | 25 | 40 | V4–V6 | 25 (palpit.) | VT, SD | Amiodarone+BB | RV | SD (autopsy) | 8 | 2/2 | |
| 146, III, 2 | c.423-1G>A | F | 10 | 0 | — | — | — | — | — | Asymptomatic | 13 | 1/0 | ||
| 151, III, 2 | c.5324G>T | R1775I | 37 | F | 37 | 30 | V1–V4 | 38 (palpit.) | VT, aSD | ICD, Disop.+BB | RV | Asymptomatic | 5 | 1/3 |
| 151, III, 4 | c.5324G>T | R1775I | F | 52 | 20 | — | — | — | — | — | Asymptomatic | 1 | 0/2 | |
| 151, IV, 1 | c.5324G>T | R1775I | M | 13 | 0 | — | — | — | — | — | Asymptomatic | 3 | 0/2 | |
| 151, IV, 4 | c.5324G>T | R1775I | M | 18 | 0 | V1 | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, I, 1 | c.3764G>A | R1255K | F | 81 | 0 | — | — | — | — | — | Asymptomatic | 1 | 0/1 | |
| 152, II, 1 | c.3764G>A | R1255K | 40 | M | 40 | 0 | V1 | 40 (aSD) | — | ICD, sotalol | — | Asymptomatic | 2 | 1/3 |
AA, aminoacidic change; aSD, aborted SD; BB, beta-blockers; Disop., disopyramide; HF, heart failure; ICD, implantable cardioverter defibrillator; LV, left ventricle; Mj, major; m, minor; NSVT, non sustained ventricular tachycardia; palpit., palpitations; QRD, QRS dispersion; RV, right ventricle; SD, sudden death; VF, ventricular fibrillation; VT, sustained ventricular tachycardia.
ECG and echocardiographic features in 26 patients carrying DSP mutations
| Case | Incomplete RBBB | QRS low voltage | qS in II, III, aVF | Negative T wave in precordial leads | Ventricular arrhythmias | Epsilon wave | LP | LVEDV mL/m2 | LVEF % | LV kinetic abn | RVEDV mL/m2 | RVEF % | RV kinetic abn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 105, II, 1 | — | — | + | — | — | — | — | np | np | np | np | np | np |
| 105, II, 4 | — | + | + | V1–V5 | VT | + | + | 77 | 45 | D | 125 | 35 | D |
| 105, II, 6 | — | — | + | V1–V5 | VT | — | np | 80 | 50 | S | 120 | 40 | D |
| 105, III, 4 | — | — | + | V1–V3 | — | — | + | 42 | 70 | — | 40 | 66 | S |
| 105, III, 7 | — | — | — | — | PVCs | — | — | 54 | 62 | — | 57 | 55 | — |
| 105, III, 9 | — | — | — | — | — | — | — | 51 | 68 | — | 48 | 70 | — |
| 105, III,10 | — | — | — | V1–V3 | NSVT | — | + | 55 | 60 | S | 70 | 60 | D |
| 105, III,11 | — | + | — | V1–V5 | VF | — | + | 75 | 45 | D | 140 | 36 | D |
| 105, III,12 | — | — | — | — | — | — | — | 57 | 65 | — | 50 | 62 | — |
| 105, III,13 | — | — | + | V1–V4 | — | — | + | 53 | 62 | — | 56 | 58 | S |
| 105, III,14 | — | — | + | V1–V3 | PVCs | — | + | 66 | 58 | — | 50 | 65 | — |
| 105, IV, 3 | — | — | — | — | — | — | — | 55 | 68 | — | 48 | 65 | — |
| 105, IV, 4 | + | — | + | V1 | VF | — | — | np | np | np | np | np | np |
| 105, IV, 5 | + | — | — | V1–V3 | PVCs | — | — | 54 | 68 | — | 54 | 66 | S |
| 105, IV, 6 | — | — | — | — | — | — | — | 55 | 65 | — | 55 | 60 | — |
| 105, IV, 7 | + | + | — | V1–V3 | NSVT | — | + | 46 | 69 | — | 75 | 62 | D |
| 105, IV, 10 | — | — | — | V1–V3 | — | — | — | 68 | 65 | S | 70 | 60 | S |
| 146, II, 2 | — | — | — | — | — | — | + | 79 | 54 | S | 60 | 65 | — |
| 146, III, 1 | + | + | — | V4–V6 | VT | — | + | 80 | 50 | D | 75 | 48 | D |
| 146, III, 2 | — | — | — | — | — | — | — | 63 | 61 | — | 56 | 55 | — |
| 151, III, 2 | — | + | — | V1–V4 | VT | + | + | 50 | 63 | — | 94 | 42 | D |
| 151, III, 4 | — | — | — | — | — | — | — | 53 | 72 | — | 55 | 61 | S |
| 151, IV, 1 | — | — | — | — | — | — | + | 53 | 65 | — | 50 | 60 | — |
| 151, IV, 4 | — | — | — | V1 | — | — | — | 74 | 62 | — | 65 | 54 | — |
| 152, I, 1 | — | — | — | — | — | — | — | 62 | 69 | — | 50 | 60 | — |
| 152, II, 1 | + | — | — | V1 | VF | — | — | 61 | 65 | — | 76 | 48 | S |
| Case | Incomplete RBBB | QRS low voltage | qS in II, III, aVF | Negative T wave in precordial leads | Ventricular arrhythmias | Epsilon wave | LP | LVEDV mL/m2 | LVEF % | LV kinetic abn | RVEDV mL/m2 | RVEF % | RV kinetic abn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 105, II, 1 | — | — | + | — | — | — | — | np | np | np | np | np | np |
| 105, II, 4 | — | + | + | V1–V5 | VT | + | + | 77 | 45 | D | 125 | 35 | D |
| 105, II, 6 | — | — | + | V1–V5 | VT | — | np | 80 | 50 | S | 120 | 40 | D |
| 105, III, 4 | — | — | + | V1–V3 | — | — | + | 42 | 70 | — | 40 | 66 | S |
| 105, III, 7 | — | — | — | — | PVCs | — | — | 54 | 62 | — | 57 | 55 | — |
| 105, III, 9 | — | — | — | — | — | — | — | 51 | 68 | — | 48 | 70 | — |
| 105, III,10 | — | — | — | V1–V3 | NSVT | — | + | 55 | 60 | S | 70 | 60 | D |
| 105, III,11 | — | + | — | V1–V5 | VF | — | + | 75 | 45 | D | 140 | 36 | D |
| 105, III,12 | — | — | — | — | — | — | — | 57 | 65 | — | 50 | 62 | — |
| 105, III,13 | — | — | + | V1–V4 | — | — | + | 53 | 62 | — | 56 | 58 | S |
| 105, III,14 | — | — | + | V1–V3 | PVCs | — | + | 66 | 58 | — | 50 | 65 | — |
| 105, IV, 3 | — | — | — | — | — | — | — | 55 | 68 | — | 48 | 65 | — |
| 105, IV, 4 | + | — | + | V1 | VF | — | — | np | np | np | np | np | np |
| 105, IV, 5 | + | — | — | V1–V3 | PVCs | — | — | 54 | 68 | — | 54 | 66 | S |
| 105, IV, 6 | — | — | — | — | — | — | — | 55 | 65 | — | 55 | 60 | — |
| 105, IV, 7 | + | + | — | V1–V3 | NSVT | — | + | 46 | 69 | — | 75 | 62 | D |
| 105, IV, 10 | — | — | — | V1–V3 | — | — | — | 68 | 65 | S | 70 | 60 | S |
| 146, II, 2 | — | — | — | — | — | — | + | 79 | 54 | S | 60 | 65 | — |
| 146, III, 1 | + | + | — | V4–V6 | VT | — | + | 80 | 50 | D | 75 | 48 | D |
| 146, III, 2 | — | — | — | — | — | — | — | 63 | 61 | — | 56 | 55 | — |
| 151, III, 2 | — | + | — | V1–V4 | VT | + | + | 50 | 63 | — | 94 | 42 | D |
| 151, III, 4 | — | — | — | — | — | — | — | 53 | 72 | — | 55 | 61 | S |
| 151, IV, 1 | — | — | — | — | — | — | + | 53 | 65 | — | 50 | 60 | — |
| 151, IV, 4 | — | — | — | V1 | — | — | — | 74 | 62 | — | 65 | 54 | — |
| 152, I, 1 | — | — | — | — | — | — | — | 62 | 69 | — | 50 | 60 | — |
| 152, II, 1 | + | — | — | V1 | VF | — | — | 61 | 65 | — | 76 | 48 | S |
abn, abnormalities; D: diffuse; LP, late potentials; LVEDV, left ventricular end diastolic volume; LVEF, left ventricular ejection fraction, LV: left ventricle; np, not performed; NSVT, non sustained ventricular tachycardia; PVCs, premature ventricular complexes; RBBB, right bundle branch block; RV, right ventricle; RVEDV, right ventricular end diastolic volume; RVEF, right ventricular ejection fraction; S, segmental; VF, ventricular fibrillation; VT, sustained ventricular tachycardia.
ECG and echocardiographic features in 26 patients carrying DSP mutations
| Case | Incomplete RBBB | QRS low voltage | qS in II, III, aVF | Negative T wave in precordial leads | Ventricular arrhythmias | Epsilon wave | LP | LVEDV mL/m2 | LVEF % | LV kinetic abn | RVEDV mL/m2 | RVEF % | RV kinetic abn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 105, II, 1 | — | — | + | — | — | — | — | np | np | np | np | np | np |
| 105, II, 4 | — | + | + | V1–V5 | VT | + | + | 77 | 45 | D | 125 | 35 | D |
| 105, II, 6 | — | — | + | V1–V5 | VT | — | np | 80 | 50 | S | 120 | 40 | D |
| 105, III, 4 | — | — | + | V1–V3 | — | — | + | 42 | 70 | — | 40 | 66 | S |
| 105, III, 7 | — | — | — | — | PVCs | — | — | 54 | 62 | — | 57 | 55 | — |
| 105, III, 9 | — | — | — | — | — | — | — | 51 | 68 | — | 48 | 70 | — |
| 105, III,10 | — | — | — | V1–V3 | NSVT | — | + | 55 | 60 | S | 70 | 60 | D |
| 105, III,11 | — | + | — | V1–V5 | VF | — | + | 75 | 45 | D | 140 | 36 | D |
| 105, III,12 | — | — | — | — | — | — | — | 57 | 65 | — | 50 | 62 | — |
| 105, III,13 | — | — | + | V1–V4 | — | — | + | 53 | 62 | — | 56 | 58 | S |
| 105, III,14 | — | — | + | V1–V3 | PVCs | — | + | 66 | 58 | — | 50 | 65 | — |
| 105, IV, 3 | — | — | — | — | — | — | — | 55 | 68 | — | 48 | 65 | — |
| 105, IV, 4 | + | — | + | V1 | VF | — | — | np | np | np | np | np | np |
| 105, IV, 5 | + | — | — | V1–V3 | PVCs | — | — | 54 | 68 | — | 54 | 66 | S |
| 105, IV, 6 | — | — | — | — | — | — | — | 55 | 65 | — | 55 | 60 | — |
| 105, IV, 7 | + | + | — | V1–V3 | NSVT | — | + | 46 | 69 | — | 75 | 62 | D |
| 105, IV, 10 | — | — | — | V1–V3 | — | — | — | 68 | 65 | S | 70 | 60 | S |
| 146, II, 2 | — | — | — | — | — | — | + | 79 | 54 | S | 60 | 65 | — |
| 146, III, 1 | + | + | — | V4–V6 | VT | — | + | 80 | 50 | D | 75 | 48 | D |
| 146, III, 2 | — | — | — | — | — | — | — | 63 | 61 | — | 56 | 55 | — |
| 151, III, 2 | — | + | — | V1–V4 | VT | + | + | 50 | 63 | — | 94 | 42 | D |
| 151, III, 4 | — | — | — | — | — | — | — | 53 | 72 | — | 55 | 61 | S |
| 151, IV, 1 | — | — | — | — | — | — | + | 53 | 65 | — | 50 | 60 | — |
| 151, IV, 4 | — | — | — | V1 | — | — | — | 74 | 62 | — | 65 | 54 | — |
| 152, I, 1 | — | — | — | — | — | — | — | 62 | 69 | — | 50 | 60 | — |
| 152, II, 1 | + | — | — | V1 | VF | — | — | 61 | 65 | — | 76 | 48 | S |
| Case | Incomplete RBBB | QRS low voltage | qS in II, III, aVF | Negative T wave in precordial leads | Ventricular arrhythmias | Epsilon wave | LP | LVEDV mL/m2 | LVEF % | LV kinetic abn | RVEDV mL/m2 | RVEF % | RV kinetic abn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 105, II, 1 | — | — | + | — | — | — | — | np | np | np | np | np | np |
| 105, II, 4 | — | + | + | V1–V5 | VT | + | + | 77 | 45 | D | 125 | 35 | D |
| 105, II, 6 | — | — | + | V1–V5 | VT | — | np | 80 | 50 | S | 120 | 40 | D |
| 105, III, 4 | — | — | + | V1–V3 | — | — | + | 42 | 70 | — | 40 | 66 | S |
| 105, III, 7 | — | — | — | — | PVCs | — | — | 54 | 62 | — | 57 | 55 | — |
| 105, III, 9 | — | — | — | — | — | — | — | 51 | 68 | — | 48 | 70 | — |
| 105, III,10 | — | — | — | V1–V3 | NSVT | — | + | 55 | 60 | S | 70 | 60 | D |
| 105, III,11 | — | + | — | V1–V5 | VF | — | + | 75 | 45 | D | 140 | 36 | D |
| 105, III,12 | — | — | — | — | — | — | — | 57 | 65 | — | 50 | 62 | — |
| 105, III,13 | — | — | + | V1–V4 | — | — | + | 53 | 62 | — | 56 | 58 | S |
| 105, III,14 | — | — | + | V1–V3 | PVCs | — | + | 66 | 58 | — | 50 | 65 | — |
| 105, IV, 3 | — | — | — | — | — | — | — | 55 | 68 | — | 48 | 65 | — |
| 105, IV, 4 | + | — | + | V1 | VF | — | — | np | np | np | np | np | np |
| 105, IV, 5 | + | — | — | V1–V3 | PVCs | — | — | 54 | 68 | — | 54 | 66 | S |
| 105, IV, 6 | — | — | — | — | — | — | — | 55 | 65 | — | 55 | 60 | — |
| 105, IV, 7 | + | + | — | V1–V3 | NSVT | — | + | 46 | 69 | — | 75 | 62 | D |
| 105, IV, 10 | — | — | — | V1–V3 | — | — | — | 68 | 65 | S | 70 | 60 | S |
| 146, II, 2 | — | — | — | — | — | — | + | 79 | 54 | S | 60 | 65 | — |
| 146, III, 1 | + | + | — | V4–V6 | VT | — | + | 80 | 50 | D | 75 | 48 | D |
| 146, III, 2 | — | — | — | — | — | — | — | 63 | 61 | — | 56 | 55 | — |
| 151, III, 2 | — | + | — | V1–V4 | VT | + | + | 50 | 63 | — | 94 | 42 | D |
| 151, III, 4 | — | — | — | — | — | — | — | 53 | 72 | — | 55 | 61 | S |
| 151, IV, 1 | — | — | — | — | — | — | + | 53 | 65 | — | 50 | 60 | — |
| 151, IV, 4 | — | — | — | V1 | — | — | — | 74 | 62 | — | 65 | 54 | — |
| 152, I, 1 | — | — | — | — | — | — | — | 62 | 69 | — | 50 | 60 | — |
| 152, II, 1 | + | — | — | V1 | VF | — | — | 61 | 65 | — | 76 | 48 | S |
abn, abnormalities; D: diffuse; LP, late potentials; LVEDV, left ventricular end diastolic volume; LVEF, left ventricular ejection fraction, LV: left ventricle; np, not performed; NSVT, non sustained ventricular tachycardia; PVCs, premature ventricular complexes; RBBB, right bundle branch block; RV, right ventricle; RVEDV, right ventricular end diastolic volume; RVEF, right ventricular ejection fraction; S, segmental; VF, ventricular fibrillation; VT, sustained ventricular tachycardia.
References
Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Martini B, Stritoni P, Fasoli G. Familial occurrence of right ventricular dysplasia. A study involving nine families.
Nava A, Rossi L, Thiene G.
Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases.
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people.
Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy or myocarditis?
Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G, Scognamiglio R, Corrado D, Thiene G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease).
Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2).
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy.
Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair.
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, Macrae CA, Peters S, Grossman KS, Michely B, Sasse-Klaassen S, Birchmeier M, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy.
Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1.
Green KJ, Parry DA, Steinert PM, Virata ML, Wagner RM, Angst BD, Nilles LA. Structure of the human desmoplakins. Implications for function in the desmosomal plaque.
Leung CL, Green KJ, Liem RKH. Plakins: a family of versatile cytolinker proteins.
Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, Hughes AE. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma.
Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy.
Duran M, Avellan F, Carvajal L. Miocardiopatia dilatada en las displasias del ectodermo. Observaciones electroecocardiograficas en la hiperqueratosis palmoplantar con pelo lanoso.
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma.
Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, McLean WH, Pulkkinen L, Uitto J, Christiano AM, Eady RA, McGrath JA. Compound heterozygosity for non-sense and missense mutations in desmoplakin underlies skin fragility/woolly hair syndrome.
Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, Daliento L, Buja G, Corrado D, Danieli GA, Thiene G. Clinical profile and long term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy.
McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.
Schiller NB. Two-dimensional echocardiographic determination of left ventricular volume, systolic function, and mass. Summary and discussion of the 1989 recommendations of the American Society of Echocardiography.
Levine RA, Gibson TC, Aretz T, Gillam LD, Guyer DE, King ME, Weyman AE. Echocardiographic measurement of right ventricular volume.
Scognamiglio R, Fasoli G, Nava A, Miraglia G, Thiene G, Dalla Volta S. Contribution of cross-sectional echocardiography to the diagnosis of right ventricular dysplasia at the asymptomatic stage.
Protonotarios N, Tsatsopoulou A, Anastasakis A, Sevdalis E, McKoy G, Stratos K, Gatzoulis K, Tentolouris K, Spiliopoulou C, Panagiotakos D, McKenna W, Toutouzas P. Genotype–phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin.
McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server.
Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage.
Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, Denning MF, Green KJ. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin–plakoglobin complexes.
Kouklis PD, Hutton E, Fuchs E. Making a connection: direct binding between keratin intermediate filaments and desmosomal proteins-desmoplakin.
Protonotarios N, Tsatsopoulou A, Fontaine G. Naxos disease: keratoderma, scalp modifications and cardiomyopathy.
Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. Structural and molecular pathology of the heart in Carvajal syndrome.
Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, Sachdev B, Rowland E, Elliott PM, McKenna WJ. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}