
Abstract
Environmental risk factors have been implicated in the etiology of psychotic disorders, with growing evidence showing the adverse effects of migration, social marginalization, urbanicity, childhood trauma, social defeat, and other adverse experiences on mental health in vulnerable populations. Collectively, social stress may be one mechanism that could link these environmental risk factors. The exact mechanism(s) by which social stress can affect brain function, and in particular the molecular targets involved in psychosis (such as the dopaminergic (DA) system), is (are) not fully understood. In this review, we will discuss the interplay between social environmental risk factors and molecular changes in the human brain; in particular, we will highlight the impact of social stress on three specific neurochemical systems: DA, neuroinflammation/immune, and endocannabinoid (eCB) signaling. We have chosen the latter two molecular pathways based on emerging evidence linking schizophrenia to altered neuroinflammatory processes and cannabis use. We further identify key developmental periods in which social stress interacts with these pathways, suggesting window(s) of opportunities for novel interventions. Taken together, we suggest that they may have a key role in the pathogenesis and disease progression, possibly provide novel treatment options for schizophrenia, and perhaps even prevent it.
Similar content being viewed by others
Main
Psychosis is characterized by a constellation of symptoms that includes abnormal perceptions and beliefs, usually called positive symptoms. Negative symptoms (eg, anhedonia, social withdrawal, etc) and cognitive deficits (eg, impaired memory, attention, executive functions, etc) are also evident, and represent major predictors of functional outcome. Epidemiological data have consistently demonstrated a well-replicated association between early environmental social risk factors and psychosis. The exact mechanism(s) by which social stress can affect brain function, and in particular the molecular targets involved in psychosis (such as the dopaminergic (DA) system), are not fully understood. In this review, we will discuss the interplay between social environmental risk factors and molecular changes in the human brain; in particular, we will highlight the impact of social stress on three specific neurochemical systems: DA, neuroinflammation/immune, and endocannabinoid (eCB) signaling. We have chosen the latter two molecular pathways based on emerging evidence linking schizophrenia to altered neuroinflammatory processes (Carter et al, 2014) and cannabis use (Andreasson et al, 1987; Arseneault et al, 2002; Harley et al, 2010; Leweke et al, 2007; Morgan et al, 2013). Although a number of other neurochemical systems have been implicated in schizophrenia, such as the glutamate system (Carter et al, 2014; Coyle, 2012; Javitt, 2012), the scope of this review is limited to the molecular systems with existing human data on the effects of psychosocial stress, notwithstanding encouraging findings regarding stress-induced glutamate alterations obtained in animal studies (Gan et al, 2014; Jiang et al, 2013). In this article, stress is broadly defined as either cortisol alterations or social manipulations, which are appraised to exceed the adaptive capacity to cope.
Evidence for the association between social stress, the hypothalamus–pituitary–adrenal axis, and psychosis
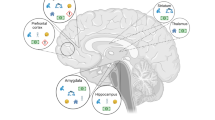
The etiology of schizophrenia is multifactorial and felt to reflect an interaction between genetic vulnerability and environmental contributors (Figure 1). Risk and protective factors, acting at a number of levels over time, appear to influence an individual’s potential for developing of psychosis. In prospective longitudinal studies, it has been observed that individuals with schizophrenia experience an increased number of stressful life events in the period immediately preceding a relapse (Nuechterlein et al, 1992; Pallanti et al, 1997). This association is not entirely consistent (Hirsch et al, 1996), but gains further support from ecologically valid studies (ie, at the time of the event) (Myin-Germeys et al, 2005). Cortisol levels, an index of stress, have been positively associated with psychotic symptoms in schizophrenia (Walder et al, 2000), although results are not entirely consistent (Mondelli et al, 2010). Differences in the level of diurnal cortisol has been reported in schizophrenia (Mondelli et al, 2010; Walder et al, 2000). For example, higher levels of diurnal cortisol levels were reported in patients on antipsychotic treatments for <2 weeks as compared with controls or patients on antipsychotic treatments for >2 weeks (Mondelli et al, 2010). In addition, hypothalamus–pituitary–adrenal (HPA) over activity has been reported in schizophrenia patients including increased adrenocorticotropic hormone response to pharmacologic/psychosocial challenges (Elman et al, 1998) (for a review see Walker and Diforio, 1997) and abnormalities in glucocorticoid receptors (Perlman et al, 2004; Webster et al, 2002). A recent major multicenter study found that, after 2 years, high baseline cortisol levels predicted transition to psychotic level symptoms in at-risk youths (Walker et al, 2013). This is in line with a recent review, which suggests that, in conjunction with genetic risk factors, childhood adversity and trauma modifies the neurodevelopmental process resulting in increased stress sensitization during adolescence and early adulthood, leading to increased risk for psychosis (Howes and Murray, 2014).

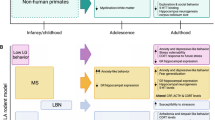
We have focused our review on three main neurochemical systems involved in psychosis: the dopaminergic (DA), endocannabinoid (eCB), and neuroinflammatory systems. However, the prominent role of the glutamatergic (Glu) system in psychosis cannot be ignored. Stage-specific stressors are indicated above the rings, whereas the big arrows represent the critical effect of social stress across all stages. The rings indicate possible environmental factors at each developmental stage that increase the risk for psychosis. At the early developmental stage, genetic vulnerability may combine with prenatal and perinatal insults (eg, maternal immune activation) to prime some individuals. In addition, social stressors experienced in childhood may lead to structural and functional development abnormalities leading to enhanced vulnerability, perhaps through environmental impact on gene expression (epigenetics). At the adolescence stage, in addition to the important social stressors typical for this age range (moving out of parental home, new schooling, peers), drug use affecting DA, Glu, and eCB signaling may amplify the premorbid neurochemical aberrancies carried over from prior stages. At all developmental stages, the genetic traits of the individual confer either resilience against or vulnerability leading to disease. A psychotic break occurs when multiple factors coalesce together, typically in early adulthood. Psychosocial or pharmacological interventions, addressing social stress and/or targeting these neurochemical systems in specific timings (such as in adolescence) may divert this trajectory away from psychosis towards resilience and health.
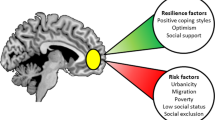
Epidemiologic studies are consistent with the key role of stress/cortisol for psychosis. For example, the most consistent findings in the epidemiology of schizophrenia is the higher incidence of the disorder among migrant and ethnic minority groups (Akdeniz et al, 2014; Boydell et al, 2001; Morgan and Fearon, 2007; van Os et al, 2001). The reasons for the increased risk of schizophrenia and other psychoses in migrant groups are not entirely clear, but among other reasons, different forms of social stress may be responsible. Urbanicity, social isolation, social defeat, disrupted familial environment in early childhood, language and cultural maladjustment, childhood abuse, and persistent experiences of victimization and discrimination in migrants are important potential factors for psychosis (Morgan and Fearon, 2007; Selten et al, 2007). Growing up in an urban environment is associated with an increased risk of developing psychosis (Krabbendam and van Os, 2005; van Os et al, 2001, 2004) and abnormal brain responses to stress in normal volunteers (Akdeniz et al, 2014; Lederbogen et al, 2011). The finding of increased likelihood of psychosis in migrants when they live in environments where they represent a minority also reflects the impact of perceived social stress (Boydell et al, 2001). Consistent with this view, social defeat has been postulated as the underlying mechanism linking psychosocial aversive events to risk for psychosis (Selten et al, 2013). In line with this, the ethnic minority status in immigrants is associated with increased cortisol levels (Squires et al, 2012), social defeat with internalized racism is associated with dysfunctional diurnal cortisol secretion (Tull et al, 2005), and differential stress-related brain responses in urban dwellers (Akdeniz et al, 2014; Lederbogen et al, 2011). Moreover, flatter and steeper changes in diurnal cortisol levels have been found in severely and moderately neglected/abused individuals, respectively (van der Vegt et al, 2009). Furthermore, pronounced reductions in hippocampal volume, a brain region that has a key role in the HPA, has been repeatedly involved in psychosis (Wright et al, 2000).
Psychosocial stress and DA signaling
The predominant biological theory of schizophrenia holds that DA hyperactivity in the striatum (eg, associative striatum) represents a neurochemical abnormality underlying positive psychotic symptoms in schizophrenia (Howes and Kapur, 2009; Laruelle and Abi-Dargham, 1999). In line with this, the diathesis-stress model suggests that the HPA axis may trigger a cascade of events resulting in neural circuit dysfunction, including alterations in DA signaling (Walker and Diforio, 1997).
Positron emission tomography (PET) allows to investigate central DA response to stress in living humans. To date, a number of PET studies have shown increased DA release following either a psychosocial or a metabolic stress task (Adler et al, 2000; Pruessner et al, 2004). Pruessner et al (2004) was the first to report increased DA release in response to a psychosocial stress task in those who reported low maternal care. Reduction in [11C]raclopride (a D2 antagonist radioligand) binding potentials in the ventral striatum relative to the non-displaceable compartment in the brain (BPND) during stress averaged ~10% (Pruessner et al, 2004), a magnitude of change of the same order as that observed after administration of amphetamine in healthy volunteers (Laruelle and Abi-Dargham, 1999). Interestingly, the amount of DA released, indexed as a reduction of [11C]raclopride binding (BPND), was proportional to the salivary cortisol response to stress (Pruessner et al, 2004). The high correlation between the two values suggests a close link between cortisol and DA stress responses. This association was also replicated in a recent study using [11C]-(+)-PHNO, a DA D2/3-specific agonist radioligand (Mizrahi et al, 2012). Adler et al (2000) has also provided further evidence, reporting that a metabolic stressor was linked to a significant reduction in striatal [11C]raclopride BPND. However, changes in DA release were not replicated in a single study, which used a different stress paradigm, with no apparent social feedback (Montgomery et al, 2006). Although this line of research is important as it links task-related changes to alterations in DA receptor binding, it does introduce the potential confound of additional head movement.
Only one study investigated DA release in response to a psychosocial stress challenge in psychosis-related disorders. More specifically, Mizrahi et al (2012) showed significant displacement of [11C]-(+)-PHNO in antipsychotic-naive patients with schizophrenia, with no effect in healthy volunteers; interestingly, an intermediate response was observed in those at elevated clinical risk for developing schizophrenia. Consistent with the PET data, the largest stress-induced changes in salivary cortisol was present in the schizophrenia group, followed by the clinical high-risk group (Mizrahi et al, 2012). Furthermore, the percent change in the cortisol response between the control and stress challenge was significantly associated with stress-induced DA release in the associative striatum (Mizrahi et al, 2012). Although these data do not prove causation, they provide the first evidence of increased DA release in response to a psychosocial stress challenge in psychosis-related disorders. This line of thinking would be consistent with recent theories of how stress may be affecting DA signaling. For example, stress is known to damage the hippocampus (Mondelli et al, 2010, 2011), a brain region commonly reported to be altered in both post-mortem (Benes, 1999) and imaging studies (Nelson et al, 1998) in schizophrenia, which was also proposed to underlie the DA hyperactivity in a well-validated animal model of schizophrenia (Lodge and Grace, 2007). In addition, preclinical studies showed a stronger connection between neonatal hippocampal injuries and increased stress-associated dopamine release (Cabungcal et al, 2014; Heinz et al, 1999). In summary, abnormal DA sensitivity in response to stress may be one pathway through which the social world interacts with biology to confer a higher risk of schizophrenia.
Other molecular mechanisms underlying psychosocial stress as a risk factor for psychosis
Immune System and Psychosis
It is increasingly recognized that other molecular mechanisms may contribute to schizophrenia, and its vulnerability. For example, several lines of evidence point to a role for neuroinflammation in the pathogenesis of schizophrenia. These were recently reviewed (Carter et al, 2014), and the findings can be summarized as follows: (a) abnormal immune activation and elevated maternal proinflammatory cytokines during pregnancy significantly linked to later development of schizophrenia in offspring; (b) elevated inflammatory proteins (eg, cytokines) in schizophrenia patients; (c) potential significant modulatory effects of antipsychotics on neuroinflammation; (d) role for anti-inflammatory agents in the treatment of schizophrenia, including its putative prodrome; (e) PET studies either detected increased neuroinflammation or in negative studies, suggesting a significant association with positive psychotic symptoms and duration of illness; (f) association between psychosis and antibodies to membrane receptors; and (g) genome-wide association studies demonstrating a significant role of major histocompatibility complex genes in schizophrenia, among other genes (Consortium SWGotPG, 2014; Stefansson et al, 2009). Taken together, these studies suggest a prominent role for neuroinflammation/immune activation in psychosis.
Psychosocial Stress and the Immune System
Different forms of social stress have been associated with inflammatory responses, although the nature of the stress (eg, acute vs chronic, social vs physical) significantly affects these interactions. The stress inflammation link has been very well characterized in previous reviews (Kiecolt-Glaser et al, 2002); however, there has been some newer studies that add to the growing interest between psychosis, stress, and neuroinflammation. For example, acute psychosocial stress elevates proinflammatory interleukins and cortisol in healthy volunteers (Yamakawa et al, 2009) in those who experience negative affect (Carroll et al, 2011), as well as individuals exposed to early life stress (Carpenter et al, 2010), whereas childhood maltreatment and being raised in a ‘harsh family’ environment (Miller and Chen, 2010) has been linked to heightened peripheral markers of inflammation (Danese et al, 2007, 2008; Dennison et al, 2012; Hepgul et al, 2012). Social hierarchy appears to have a role in how stressors are perceived. For example, individuals who place themselves lower on the social ladder or feel lonelier demonstrate larger peripheral interleukin responses (eg, IL-6, CRP) when confronted with a psychosocial stress task vs those who place themselves higher on the social ladder (Coelho et al, 2014; Jaremka et al, 2013). Unfortunately, as of yet no study has investigated the role of psychosocial stress on the immune response in patients with schizophrenia-related disorders.
In preclinical studies, stress (glucocorticoid administration) induces activation of inflammatory responses in activated microglia (Frank et al, 2010) and, conversely, inflammatory cytokines can disrupt glucocorticoid receptor function (Pace and Miller, 2009). For example, stress-induced alterations in the glucocorticoid receptor can disrupt the well-recognized ability of glucocorticoids to restrain the inflammatory response (Rhen and Cidlowski, 2005). In a recent study, prenatal immune activation and peripubertal stress produced synergistic effects in the development of key sensorimotor gating deficiencies such as prepulse inhibition and acoustic startle reflex, both considered viable (albeit deficient) animal models of schizophrenia. Later applications of stress (ie, not peripubertal but in adulthood) do not elicit these same alterations, underscoring the precise timing of the postnatal stress challenge vis-á-vis its interaction with the prenatal immune system. Taken together, prenatal immune activation and peripubertal stress lead to enhanced DA levels in the hippocampus, increased expression of markers of activated microglia in the hippocampus and prefrontal cortex, and elevated levels of proinflammatory cytokines (Giovanoli et al, 2013). In summary, neuroinflammation would thus appear to be another molecular pathway through which social stress can affect neurochemistry and alter brain function/structure in a manner that increases risk for schizophrenia.
Cannabinoids and Psychosis
Cannabinoids, the active components of cannabis, exert their effects on the brain by acting on the eCB system. The eCB comprises the enzymes involved in their synthesis (eg, diacylglycerol lipases) and inactivation (eg, fatty acid amide hydrolase (FAAH); monoacylglycerol lipase), as well as the receptors (eg, CB1, CB2) that mediate physiological effects of the eCBs (anandamide (AEA) and 2-arachidonoylglycerol) (Di Marzo et al, 1994). Among other functions, the eCB system is involved in neuroprotection, modulation of nociception, control of certain phases of memory processing, modulation of immune and inflammatory responses, and appetite regulation (Di Marzo et al, 1994). eCB synaptic signaling works in a retrograde manner (Wang and Ueda, 2009).The lipophilic eCBs, including AEA and 2AG, are synthesized ‘on demand’ in the postsynaptic neuron and released into the synapse without any vesicular storage (Wang and Ueda, 2009). These molecules then bind to G-protein-coupled CB1 receptors in the presynaptic neuron (Kreitzer and Regehr, 2002) to modulate neurotransmitter release.
Over the years, a number of separate lines of research have converged on cannabinoids, including eCBs, as key contributors to schizophrenia: (a) ~2-fold increase in the incidence of schizophrenia with early cannabis use (Andreasson et al, 1987); (b) elevated levels of eCBs in cerebrospinal fluid (CSF) of patients with schizophrenia, including marked (up to eightfold) elevations of AEA (Leweke et al, 2007); (c) reduced peripheral expression of eCB synthesis enzymes and increased expression of degradative enzymes in first episode schizophrenia (Bioque et al, 2013); (d) potential association between CB1 receptors polymorphism (CNR1) and schizophrenia (Ujike et al, 2002); (e) increased CB1 binding in the dorsolateral prefrontal cortex (Jenko et al, 2012) and anterior cingulate cortex based on post-mortem autoradiography studies (Zavitsanou et al, 2004); and (e) elevated CB1 binding in vivo in patients with schizophrenia, as measured by two PET studies (Ceccarini et al, 2013; Wong et al, 2010). Although still in its infancy, these studies sometimes, with inconsistent findings, point to a prominent role of eCB in schizophrenia.
Psychosocial Stress and eCB
The presence of CB1 receptors within the corticolimbic circuits regulating the HPA axis, in combination with the stress-reducing properties reported by cannabis users, suggests a role for the eCB system in stress regulation. Animal studies have provided compelling evidence implicating the eCBs and, in particular, AEA in the regulation of the HPA axis (Hill et al, 2005, 2009; Rademacher et al, 2008). For example, both acute and repeated restraint stress increase FAAH hydrolytic activity and decrease AEA (Hill et al, 2009, 2013; Rademacher et al, 2008). The main site providing excitatory drive to the HPA axis is the amygdala (Herman et al, 2005), which regulates the extent of HPA axis responses to stressful stimuli (Hill et al, 2009). Tonic AEA release from the amygdala activates CB1 receptors, which decreases glutamate release and ultimately dampens excitatory afferents to the amygdala (Hill et al, 2009) and disinhibits it. These studies suggest that the eCB system is well implicated in the stress response. At a functional level, there is growing interest in the putative interactions between the HPA and eCBs. More specifically, it has been suggested that glucocorticoids recruit eCB in the amygdala to consolidate and store memory (Atsak et al, 2012; Campolongo et al, 2009). In addition to contributing to the adaptation of the HPA axis to stressful stimuli, eCB signaling in the amygdala also appears to be important in behavioral adaptation to aversive stimuli. For example, mice lacking the CB1 receptor exhibit prolonged expression of fear behaviors (Marsicano et al, 2002). This suggests that the eCB signaling system is an important safeguard against the effects of stress and the physiological aspects of the stress response. Interestingly, cannabinoids elicit behavioral as well as neurochemical changes that are dependent on the environmental conditions under which they are administered. For example, Δ9-tetrahydrocannabinol (THC) administered to rats housed in stressful conditions increase striatal DA uptake and metabolism, whereas such an effect is absent in rats housed under normal conditions (Littleton et al, 1976; MacLean and Littleton, 1977). Further to this point, cross-sensitization between THC and stress has been reported (Suplita et al, 2008), suggesting that the physiological and psychological effects of cannabis may be altered in individuals experiencing environmental adversity. We recently investigated potential cross-sensitization between stress and cannabis in those at risk of developing schizophrenia, showing an absence of increased DA release in response to a psychosocial stress task despite increased positive attenuated psychotic symptoms (Mizrahi et al, 2014). Such results are in line with studies involving patients with schizophrenia and concurrent substance use (Thompson et al, 2013), as well as cannabis users with psychotic experiences (Bloomfield et al, 2014), and suggest that even minute increases in DA release lead to increased psychotic-like experiences in these populations. A potential explanation comes from animal studies showing postsynaptic supersensitive DA D2 receptors in conjunction with drug use (Ginovart et al, 2012). Substance abuse frequently begins before the first psychiatric episode in schizophrenia (Hafner et al, 2013). Hafner et al (2013) demonstrated that cannabis abuse reduced the mean age of schizophrenia onset (17.7 years) as compared with patients without misuse (25.7 years). Patients with schizophrenia and those at risk for the illness exhibit alarmingly high levels of drug use, most commonly cannabis (Fowler et al, 1998; Regier et al, 1990), despite increased risk of psychotic experiences. This may be due to the incentive sensitization theory of addiction where continued drug consumption may cause a drug-induced increased DA sensitivity (Heinz, 2002; Robinson and Berridge, 1993). Of note, risk of psychosis has been shown to increase with childhood trauma and cannabis use through a synergistic interaction (Harley et al, 2010). Both chronic drug use and chronic stress can disrupt the eCB system leading to an impaired response to stress (Popoli et al, 2011). It is conceivable that social stress in vulnerable individuals leads to a dampened eCB system, further increasing cannabis use to potentially regulate the abnormal stress response; in turn, stimulation of CB1 receptors by exogenous cannabis may suppress the HPA response to stress (Mizrahi et al, 2014), thereby providing an ‘external buffer’. However, further studies are needed to understand the role of cannabis use on HPA axis and its relevance for psychosis and psychosis risk.
Although this hypothesis may be a reasonable explanation for the observed elevated cannabis use in those at risk and in patients with psychosis, it does not explain why cannabis use itself may lead to psychosis in at-risk populations. There is no data as of yet to answer this question. In fact, our understanding of eCB in psychosis and in cannabis users is very limited. While high AEA levels have been reported in the CSF of patients with schizophrenia and putative prodromal states, these are negatively associated with psychotic symptoms. In those at risk for psychosis, those with low AEA levels were more likely to transition earlier to psychosis (Koethe et al, 2009). Further, heavy cannabis use was associated with lower CSF AEA and negatively correlated with (cannabis-free) psychotic symptoms (Morgan et al, 2013). No doubt that investigating the eCB in living human brain of patients with psychosis and psychosis risk with and without cannabis use is needed. Notably, the eCB system undergoes marked changes in early life and adolescence until early adulthood (Long et al, 2012). These early life changes could potentially explain the sensitivity of this age group to cannabis use and social stress on those vulnerable for schizophrenia. In line with this, it has been shown that early cannabis use (before 16 years old) leads to increased risk for psychosis (Arseneault et al, 2002), and animal studies suggest a critical effect in this particular time period (Cass et al, 2014). In this regard, understanding the role of the eCB system, particularly during adolescence, may provide novel targets for stress regulation in at-risk populations.
Towards an integrated view of early molecular changes occurring during social stress and psychosis risk
Advances in our understanding of schizophrenia underscore both its complexity and heterogeneous nature. Given that the illness does not routinely declare itself until late adolescence/early adulthood, numerous influences may have a contributory role during this interval, reflected across presentation, symptomatology, illness trajectory, and response to treatment (Case et al, 2011).
In this regard, very little is known about how the DA system, eCB, and neuroinflammatory pathways affect clinical presentation, trajectory, and response to treatment. Although elevated DA response to amphetamine has been reliably associated with both positive psychotic symptoms and antipsychotic response (Abi-Dargham et al, 2009; Laruelle and Abi-Dargham, 1999), no clinical picture is defined following eCB or inflammatory markers alterations. It may well be that enhanced neuroinflammation in psychosis could be particularly relevant to cognitive and depressive and/or negative symptoms. Notably, neuroinflammation is proposed in the pathogenesis of major depression (for a review see Miller et al, 2009), and is associated with significant risk of suicide attempts (Bottlender et al, 2000; Harvey et al, 2008). Thus, indirect evidence suggests a possible role of neuroinflammation in depressive symptoms in psychosis. Only two studies examined the relationship between C-reactive protein and cognitive symptoms in schizophrenia and both found lower scores on Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) (Dickerson et al, 2007, 2012). In addition, increased S100B levels showed impaired performance on auditory verbal learning test in schizophrenia patients (Pedersen et al, 2008). Collectively, these data suggest a possible role of neuroinflammation in cognitive and depressive symptoms, and perhaps even priming the immune system for vulnerability to stress (Giovanoli et al, 2013).
The role of eCB in psychosis clinical picture, trajectory, and response to treatment is unclear, given the paucity of data in this regard. Although still in its infancy, eCB research has suggested that it is likely that the eCB will show a prominent role in stress regulation, a line of research that needs further investigation in CHR as well as first episode psychosis patients. Because the eCB system also has a critical role in the modulation of neurotransmitter release, including dopamine and glutamate via activation of the CB1 cannabinoid receptor (Prescot et al, 2013), it is well positioned in the central nervous system to exert presynaptic inhibition of synaptic transmission at both excitatory and inhibitory synapses (Harkany et al, 2008).
In this regard, the use of clinical presentation/brain changes rather than diagnostic groups (as suggested by the NIMH Research Domain Criteria (RDOC)) would prove to be more beneficial, such that each neurochemical alterations (DA, immune, and eCB) is tapped into a particular symptoms domain and/or cluster of brain structural/functional alterations. Explorations for correlations of neurochemical alterations with cognitive/behavioral symptoms provide the opportunity to uncover novel subgroups that feature prominent phenotypes reducing heterogeneity, likely increasing response to more specific treatments. Furthermore, the potential joint alteration of these three systems has never been explored in patients with psychosis, except for the first report of Perkins et al (2015) reporting both immune and HPA axis dysregulation that jointly seem to predict psychosis transition. In summary, similar studies are needed where more than one system is interrogated. Although this will likely increase sample sizes and complexities in data analysis, multisite studies and collaborations among scientists may be able to bridge this gap. Finally, these three systems could be used to stratify participants for clinical trials, for psychosis conversion, and response to treatments. In the search for new treatment options, establishing reliable altered biomarkers represents an important step in facilitating the evaluation of new molecular compounds. Finally, because interventions, pharmacological and/or psychosocial, seem exquisitely sensitive to time (ie, in adolescence vs adulthood), future studies should as well emphasize carefully selected populations that address both age and social environment.
Research of this sort faces considerable challenges. Imaging approaches appropriate for investigating these novel neurochemical systems in humans in vivo are in the early stage of development, with specific PET radioligands that target microglial activation/neuroinflammation and eCB only becoming available recently (Damont et al, 2013; Horti et al, 2006). Furthermore, these three systems are sensitive to multiple environmental perturbations, including social stress and drug use, making measurements in clinical populations a challenge. The complexity of the temporal aspect of the progression towards disease and capturing the ‘critical window’, with some biomarkers putatively not appearing until very late in the course to disease, whereas others potentially being present from very early (ie, childhood) may also hinder potential novel interventions. Additionally, the DA, eCB, and inflammatory pathways addressed in this review are not the only molecular aspects of psychosis. Finally, new data demonstrate the complex interplay of these systems (Katona and Freund, 2008), making it difficult to tease apart the independent contributions of each in clinical studies. This could, although, also be seen as an advantage in that close links between these systems suggest the possibility to target one, whereas at the same time influencing the other. For example, cannabidiol, a major component of cannabis, has been reported to have an antipsychotic effect (Leweke et al, 2012). Whether this is due to its potential anti-inflammatory properties (Carrier et al, 2006) or its putative FAAH blockage role is currently unknown (Robson et al, 2014). Nevertheless, it provides an example of a compound that could target more than one system at the same time. Furthermore, preclinical studies that elucidate the role of social stress on these molecular pathways, and their interactions including causation/directionality would become key to truly understand these complexities.
Currently, the most widely available treatment for schizophrenia is antipsychotic medication: compounds developed for their effects on neurotransmitters, in particular dopamine. Cognitive behavioral trials have shown provocative findings in those at elevated risk for psychosis (Stafford et al, 2013), but space remains to investigate the potential effects of these and other novel psychosocial interventions targeting social stress. Based on the evidence detailed here, eCB signaling and neuroinflammation may well represent valuable new targets for drug development, notwithstanding the aforementioned challenges. Within a framework that incorporates these novel neurochemical processes, opportunities arise for non-pharmacological strategies to address the significant impact of social stress. Focusing on adolescents with multiple risk factors as a starting point, social programs addressing empowerment and resilience, drug use treatment, immigrants’ supports to reduce discrimination and marginalization, cognitive training, and so on could impact the way schizophrenia is managed within the society. Investigating how these potential factors affect molecular targets in brain may provide novel ways to treat schizophrenia, perhaps even preventing it.
Funding and disclosure
Dr Mizrahi reported no biomedical financial interests or potential conflict of interest.
References
Abi-Dargham A, van de Giessen E, Slifstein M, Kegeles LS, Laruelle M (2009). Baseline and amphetamine-stimulated dopamine activity are related in drug-naïve schizophrenic subjects. Biol Psychiatry 65: 1091–1093.
Adler CM, Elman I, Weisenfeld N, Kestler L, Pickar D, Breier A (2000). Effects of acute metabolic stress on striatal dopamine release in healthy volunteers. Neuropsychopharmacology 22: 545–550.
Akdeniz C, Tost H, Streit F et al (2014). Neuroimaging evidence for a role of neural social stress processing in ethnic minority-associated environmental risk. JAMA Psychiatry 71: 672–680.
Andreasson S, Allebeck P, Engstrom A, Rydberg U (1987). Cannabis and schizophrenia. A longitudinal study of Swedish conscripts. Lancet 2: 1483–1486.
Arseneault L, Cannon M, Poulton R, Murray R, Caspi A, Moffitt TE (2002). Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. BMJ 325: 1212–1213.
Atsak P, Roozendaal B, Campolongo P (2012). Role of the endocannabinoid system in regulating glucocorticoid effects on memory for emotional experiences. Neuroscience 204: 104–116.
Benes FM (1999). Evidence for altered trisynaptic circuitry in schizophrenic hippocampus. Biol Psychiatry 46: 589–599.
Bioque M, Garcia-Bueno B, Macdowell KS, Meseguer A, Saiz PA, Parellada M et al (2013). Peripheral endocannabinoid system dysregulation in first-episode psychosis. Neuropsychopharmacology 38: 2568–2577.
Bloomfield MA, Morgan CJ, Egerton A, Kapur S, Curran HV, Howes OD (2014). Dopaminergic function in cannabis users and its relationship to cannabis-induced psychotic symptoms. Biol Psychiatry 75: 470–478.
Bottlender R, Strauss A, Moller HJ (2000). Prevalence and background factors of depression in first admitted schizophrenic patients. Acta Psychiatr Scand 101: 153–160.
Boydell J, van Os J, McKenzie K, Allardyce J, Goel R, McCreadie RG et al (2001). Incidence of schizophrenia in ethnic minorities in London: ecological study into interactions with environment. BMJ 323: 1336–1338.
Cabungcal JH, Counotte DS, Lewis EM, Tejeda HA, Piantadosi P, Pollock C et al (2014). Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 83: 1073–1084.
Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL et al (2009). Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci USA 106: 4888–4893.
Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH (2010). Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology 35: 2617–2623.
Carrier EJ, Auchampach JA, Hillard CJ (2006). Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci USA 103: 7895–7900.
Carroll JE, Low CA, Prather AA, Cohen S, Fury JM, Ross DC et al (2011). Negative affective responses to a speech task predict changes in interleukin (IL)-6. Brain Behav Immun 25: 232–238.
Carter CS, Bullmore ET, Harrison P (2014). Is there a flame in the brain in psychosis? Biol Psychiatry 75: 258–259.
Case M, Stauffer VL, Ascher-Svanum H, Conley R, Kapur S, Kane JM et al (2011). The heterogeneity of antipsychotic response in the treatment of schizophrenia. Psychol Med 41: 1291–1300.
Cass DK, Flores-Barrera E, Thomases DR, Vital WF, Caballero A, Tseng KY (2014). CB1 cannabinoid receptor stimulation during adolescence impairs the maturation of GABA function in the adult rat prefrontal cortex. Mol Psychiatry 19: 536–543.
Ceccarini J, De Hert M, Van Winkel R, Peuskens J, Bormans G, Kranaster L et al (2013). Increased ventral striatal CB1 receptor binding is related to negative symptoms in drug-free patients with schizophrenia. Neuroimage 79: 304–312.
Coelho R, Viola TW, Walss-Bass C, Brietzke E, Grassi-Oliveira R (2014). Childhood maltreatment and inflammatory markers: a systematic review. Acta Psychiatr Scand 129: 180–192.
Consortium SWGotPG (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–427.
Coyle JT (2012). NMDA receptor and schizophrenia: a brief history. Schizophr Bull 38: 920–926.
Damont A, Roeda D, Dolle F (2013). The potential of carbon-11 and fluorine-18 chemistry: illustration through the development of positron emission tomography radioligands targeting the translocator protein 18 kDa. J Labelled Comp Radiopharm 56: 96–104.
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A (2008). Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 65: 409–415.
Danese A, Pariante CM, Caspi A, Taylor A, Poulton R (2007). Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci USA 104: 1319–1324.
Dennison U, McKernan D, Cryan J, Dinan T (2012). Schizophrenia patients with a history of childhood trauma have a pro-inflammatory phenotype. Psychol Med 42: 1865–1871.
Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC et al (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372: 686–691.
Dickerson F, Stallings C, Origoni A, Boronow J, Yolken R (2007). C-reactive protein is associated with the severity of cognitive impairment but not of psychiatric symptoms in individuals with schizophrenia. Schizophr Res 93: 261–265.
Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Yolken R (2012). Additive effects of elevated C-reactive protein and exposure to herpes simplex virus type 1 on cognitive impairment in individuals with schizophrenia. Schizophr Res 134: 83–88.
Elman I, Adler CM, Malhotra AK, Bir C, Pickar D, Breier A (1998). Effect of acute metabolic stress on pituitary–adrenal axis activation in patients with schizophrenia. Am J Psychiatry 155: 979–981.
Fowler IL, Carr VJ, Carter NT, Lewin TJ (1998). Patterns of current and lifetime substance use in schizophrenia. Schizophr Bull 24: 443–455.
Frank MG, Miguel ZD, Watkins LR, Maier SF (2010). Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun 24: 19–30.
Gan JO, Bowline E, Lourenco FS, Pickel VM (2014). Adolescent social isolation enhances the plasmalemmal density of NMDA NR1 subunits in dendritic spines of principal neurons in the basolateral amygdala of adult mice. Neuroscience 258: 174–183.
Ginovart N, Tournier BB, Moulin-Sallanon M, Steimer T, Ibanez V, Millet P (2012). Chronic Delta(9)-tetrahydrocannabinol exposure induces a sensitization of dopamine D(2)/(3) receptors in the mesoaccumbens and nigrostriatal systems. Neuropsychopharmacology 37: 2355–2367.
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R et al (2013). Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 339: 1095–1099.
Hafner H, Maurer K, an der Heiden W (2013). ABC Schizophrenia study: an overview of results since 1996. Soc Psychiatry Psychiatric Epidemiol 48: 1021–1031.
Harkany T, Mackie K, Doherty P (2008). Wiring and firing neuronal networks: endocannabinoids take center stage. Curr Opin Neurobiol 18: 338–345.
Harley M, Kelleher I, Clarke M, Lynch F, Arseneault L, Connor D et al (2010). Cannabis use and childhood trauma interact additively to increase the risk of psychotic symptoms in adolescence. Psychol Med 40: 1627–1634.
Harvey SB, Dean K, Morgan C, Walsh E, Demjaha A, Dazzan P et al (2008). Self-harm in first-episode psychosis. Br J Psychiatry 192: 178–184.
Heinz A (2002). Dopaminergic dysfunction in alcoholism and schizophrenia—psychopathological and behavioral correlates. Eur Psychiatry 17: 9–16.
Heinz A, Saunders RC, Kolachana BS, Jones DW, Gorey JG, Bachevalier J et al (1999). Striatal dopamine receptors and transporters in monkeys with neonatal temporal limbic damage. Synapse 32: 71–79.
Hepgul N, Pariante CM, Dipasquale S, DiForti M, Taylor H, Marques TR et al (2012). Childhood maltreatment is associated with increased body mass index and increased C-reactive protein levels in first-episode psychosis patients. Psychol Med 42: 1893–1901.
Herman JP, Ostrander MM, Mueller NK, Figueiredo H (2005). Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry 29: 1201–1213.
Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM et al (2013). Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry 18: 1125–1135.
Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ et al (2009). Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 34: 2733–2745.
Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ et al (2005). Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology 30: 508–515.
Hirsch S, Bowen J, Emami J, Cramer P, Jolley A, Haw C et al (1996). A one year prospective study of the effect of life events and medication in the aetiology of schizophrenic relapse. Br J Psychiatry 168: 49–56.
Horti AG, Fan H, Kuwabara H, Hilton J, Ravert HT, Holt DP et al (2006). 11C-JHU75528: a radiotracer for PET imaging of CB1 cannabinoid receptors. J Nucl Med 47: 1689–1696.
Howes OD, Kapur S (2009). The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull 35: 549–562.
Howes OD, Murray RM (2014). Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet 383: 1677–1687.
Jaremka LM, Fagundes CP, Peng J, Bennett JM, Glaser R, Malarkey WB et al (2013). Loneliness promotes inflammation during acute stress. Psychol Sci 24: 1089–1097.
Javitt DC (2012). Twenty-five years of glutamate in schizophrenia: are we there yet? Schizophr Bull 38: 911–913.
Jenko KJ, Hirvonen J, Henter ID, Anderson KB, Zoghbi SS, Hyde TM et al (2012). Binding of a tritiated inverse agonist to cannabinoid CB1 receptors is increased in patients with schizophrenia. Schizophr Res 141: 185–188.
Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K (2013). Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol Psychiatry 73: 1024–1034.
Katona I, Freund TF (2008). Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med 14: 923–930.
Kiecolt-Glaser JK, McGuire L, Robles TF, Glaser R (2002). Psychoneuroimmunology: psychological influences on immune function and health. J Consult Clin Psychol 70: 537–547.
Koethe D, Giuffrida A, Schreiber D, Hellmich M, Schultze-Lutter F, Ruhrmann S et al (2009). Anandamide elevation in cerebrospinal fluid in initial prodromal states of psychosis. Br J Psychiatry 194: 371–372.
Krabbendam L, van Os J (2005). Schizophrenia and urbanicity: a major environmental influence—conditional on genetic risk. Schizophr Bull 31: 795–799.
Kreitzer AC, Regehr WG (2002). Retrograde signaling by endocannabinoids. Curr Opin Neurobiol 12: 324–330.
Laruelle M, Abi-Dargham A (1999). Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol 13: 358–371.
Lederbogen F, Kirsch P, Haddad L, Streit F, Tost H, Schuch P et al (2011). City living and urban upbringing affect neural social stress processing in humans. Nature 474: 498–501.
Leweke FM, Giuffrida A, Koethe D, Schreiber D, Nolden BM, Kranaster L et al (2007). Anandamide levels in cerebrospinal fluid of first-episode schizophrenic patients: impact of cannabis use. Schizophr Res 94: 29–36.
Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C et al (2012). Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2: e94.
Littleton JM, Maclean KI, Brownlee G (1976). Proceedings: alterations in dopamine uptake in rat corpus striatum induced by combinations of stress and delta8-tetrahydrocannabinol (delta8-THC). Br J Pharmacol 56: 370P.
Lodge DJ, Grace AA (2007). Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci 27: 11424–11430.
Long LE, Lind J, Webster M, Weickert CS (2012). Developmental trajectory of the endocannabinoid system in human dorsolateral prefrontal cortex. BMC Neurosci 13: 87.
MacLean KI, Littleton JM (1977). Environmental stress as a factor in the response of rat brain catecholamine metabolism to delta8-tetrahydrocannabinol. Eur J Pharmacol 41: 171–182.
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG et al (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530–534.
Miller AH, Maletic V, Raison CL (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65: 732–741.
Miller GE, Chen E (2010). Harsh family climate in early life presages the emergence of a proinflammatory phenotype in adolescence. Psychol Sci 21: 848–856.
Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I et al (2012). Increased stress-induced dopamine release in psychosis. Biol Psychiatry 71: 561–567.
Mizrahi R, Kenk M, Suridjan I, Boileau I, George TP, McKenzie K et al (2014). Stress-induced dopamine response in subjects at clinical high risk for schizophrenia with and without concurrent cannabis use. Neuropsychopharmacology 39: 1479–1489.
Mondelli V, Cattaneo A, Belvederi Murri M, Di Forti M, Handley R, Hepgul N et al (2011). Stress and inflammation reduce brain-derived neurotrophic factor expression in first-episode psychosis: a pathway to smaller hippocampal volume. J Clin Psychiatry 72: 1677–1684.
Mondelli V, Dazzan P, Hepgul N, Di Forti M, Aas M, D'Albenzio A et al (2010). Abnormal cortisol levels during the day and cortisol awakening response in first-episode psychosis: the role of stress and of antipsychotic treatment. Schizophr Res 116: 234–242.
Montgomery AJ, Mehta MA, Grasby PM (2006). Is psychological stress in man associated with increased striatal dopamine levels?: A [11C]raclopride PET study. Synapse 60: 124–131.
Morgan C, Fearon P (2007). Social experience and psychosis insights from studies of migrant and ethnic minority groups. Epidemiol Psichiatr Soc 16: 118–123.
Morgan CJ, Page E, Schaefer C, Chatten K, Manocha A, Gulati S et al (2013). Cerebrospinal fluid anandamide levels, cannabis use and psychotic-like symptoms. Br J Psychiatry 202: 381–382.
Myin-Germeys I, Marcelis M, Krabbendam L, Delespaul P, van Os J (2005). Subtle fluctuations in psychotic phenomena as functional states of abnormal dopamine reactivity in individuals at risk. Biol Psychiatry 58: 105–110.
Nelson MD, Saykin AJ, Flashman LA, Riordan HJ (1998). Hippocampal volume reduction in schizophrenia as assessed by magnetic resonance imaging. Archiv Gen Psychiatry 55: 433.
Nuechterlein KH, Dawson ME, Gitlin M, Ventura J, Goldstein MJ, Snyder KS et al (1992). Developmental processes in schizophrenic disorders: longitudinal studies of vulnerability and stress. Schizophr Bull 18: 387–425.
Pace TW, Miller AH (2009). Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci 1179: 86–105.
Pallanti S, Quercioli L, Pazzagli A (1997). Relapse in young paranoid schizophrenic patients: a prospective study of stressful life events, P300 measures, and coping. Am J Psychiatry 154: 792–798.
Pedersen A, Diedrich M, Kaestner F, Koelkebeck K, Ohrmann P, Ponath G et al (2008). Memory impairment correlates with increased S100B serum concentrations in patients with chronic schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 32: 1789–1792.
Perlman WR, Webster MJ, Kleinman JE, Weickert CS (2004). Reduced glucocorticoid and estrogen receptor alpha messenger ribonucleic acid levels in the amygdala of patients with major mental illness. Biol Psychiatry 56: 844–852.
Perkins DO, Jeffries CD, Addington J, Bearden CE, Cadenhead KS et al (2015). Towards a psychosis risk blood diagnostic for persons experiencing high-risk symptoms: preliminary results from the NAPLS project. Schizophr Bull 41: 419–428.
Popoli M, Yan Z, McEwen B, Sanacora G (2011). The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 13: 22–37.
Prescot AP, Renshaw PF, Yurgelun-Todd DA (2013). Gamma-amino butyric acid and glutamate abnormalities in adolescent chronic marijuana smokers. Drug Alcohol Depend 129: 232–239.
Pruessner JC, Champagne F, Meaney MJ, Dagher A (2004). Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci 24: 2825–2831.
Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, Hillard CJ (2008). Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology 54: 108–116.
Regier DA, Farmer ME, Rae DS, Locke BZ, Keith SJ, Judd LL et al (1990). Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 264: 2511–2518.
Rhen T, Cidlowski JA (2005). Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353: 1711–1723.
Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev 18: 247–291.
Robson PJ, Guy GW, Di Marzo V (2014). Cannabinoids and schizophrenia: therapeutic prospects. Curr Pharmaceut Des 20: 2194–2204.
Selten JP, Cantor-Graae E, Kahn RS (2007). Migration and schizophrenia. Curr Opin Psychiatry 20: 111–115.
Selten JP, van der Ven E, Rutten BP, Cantor-Graae E (2013). The social defeat hypothesis of schizophrenia: an update. Schizophr Bull 39: 1180–1186.
Squires EC, McClure HH, Martinez CR Jr, Eddy JM, Jimenez RA, Isiordia LE et al (2012). Diurnal cortisol rhythms among Latino immigrants in Oregon, USA. J Physiol Anthropol 31: 19.
Stafford MR, Jackson H, Mayo-Wilson E, Morrison AP, Kendall T (2013). Early interventions to prevent psychosis: systematic review and meta-analysis. BMJ 346: f185.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D et al (2009). Common variants conferring risk of schizophrenia. Nature 460: 744–747.
Suplita RL 2nd, Eisenstein SA, Neely MH, Moise AM, Hohmann AG (2008). Cross-sensitization and cross-tolerance between exogenous cannabinoid antinociception and endocannabinoid-mediated stress-induced analgesia. Neuropharmacology 54: 161–171.
Thompson JL, Urban N, Slifstein M, Xu X, Kegeles LS, Girgis RR et al (2013). Striatal dopamine release in schizophrenia comorbid with substance dependence. Mol Psychiatry 18: 909–915.
Tull ES, Sheu Y-T, Butler C, Cornelious K (2005). Relationships between perceived stress, coping behavior and cortisol secretion in women with high and low levels of internalized racism. J Natl Med Assoc 97: 206–212.
Ujike H, Takaki M, Nakata K, Tanaka Y, Takeda T, Kodama M et al (2002). CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Mol Psychiatry 7: 515–518.
van der Vegt EJ, van der Ende J, Kirschbaum C, Verhulst FC, Tiemeier H (2009). Early neglect and abuse predict diurnal cortisol patterns in adults A study of international adoptees. Psychoneuroendocrinology 34: 660–669.
van Os J, Hanssen M, Bijl RV, Vollebergh W (2001). Prevalence of psychotic disorder and community level of psychotic symptoms: an urban-rural comparison. Arch Gen Psychiatry 58: 663–668.
van Os J, Pedersen CB, Mortensen PB (2004). Confirmation of synergy between urbanicity and familial liability in the causation of psychosis. Am J Psychiatry 161: 2312–2314.
Walder DJ, Walker EF, Lewine RJ (2000). Cognitive functioning, cortisol release, and symptom severity in patients with schizophrenia. Biol Psychiatry 48: 1121–1132.
Walker EF, Diforio D (1997). Schizophrenia: a neural diathesis-stress model. Psychol Rev 104: 667–685.
Walker EF, Trotman HD, Pearce BD, Addington J, Cadenhead KS, Cornblatt BA et al (2013). Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biol Psychiatry 74: 410–417.
Wang J, Ueda N (2009). Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat 89: 112–119.
Webster MJ, Knable MB, O'Grady J, Orthmann J, Weickert CS (2002). Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry 7: 985–994 924.
Wong DF, Kuwabara H, Horti AG, Raymont V, Brasic J, Guevara M et al (2010). Quantification of cerebral cannabinoid receptors subtype 1 (CB1) in healthy subjects and schizophrenia by the novel PET radioligand [11C]OMAR. Neuroimage 52: 1505–1513.
Wright IC, Rabe-Hesketh S, Woodruff PW, David AS, Murray RM, Bullmore ET (2000). Meta-analysis of regional brain volumes in schizophrenia. Am J Psychiatry 157: 16–25.
Yamakawa K, Matsunaga M, Isowa T, Kimura K, Kasugai K, Yoneda M et al (2009). Transient responses of inflammatory cytokines in acute stress. Biol Psychol 82: 25–32.
Zavitsanou K, Garrick T, Huang XF (2004). Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 28: 355–360.
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (Canada), National Institutes of Mental Health (R01MH100043 United States), and CAMH Foundation. We thanks Drs Gary Remington and Alan Wilson for the intellectual environment in which these thoughts were conceived and nurtured Drs Gary Remington, Miran Kenk, and Ginnie Wilson for comments about the style of this article; and Aaron Prosser for help with the figure.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Mizrahi, R. Social Stress and Psychosis Risk: Common Neurochemical Substrates?. Neuropsychopharmacol 41, 666–674 (2016). https://doi.org/10.1038/npp.2015.274
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.274
This article is cited by
-
The association between psychosocial stress, interpersonal sensitivity, social withdrawal and psychosis relapse: a systematic review
Schizophrenia (2023)
-
Stress symptoms and associated factors among adolescents in Dhaka, Bangladesh: findings from a cross-sectional study
BMC Psychiatry (2022)
-
Altered relationship between cortisol response to social stress and mediotemporal function during fear processing in people at clinical high risk for psychosis: a preliminary report
European Archives of Psychiatry and Clinical Neuroscience (2022)
-
Striatal dopamine synthesis capacity in autism spectrum disorder and its relation with social defeat: an [18F]-FDOPA PET/CT study
Translational Psychiatry (2021)
-
Urbanicity, hypothalamic-pituitary-adrenal axis functioning, and behavioral and emotional problems in children: a path analysis
BMC Psychology (2020)
