
Abstract
Neuropsychiatric disorders vary substantially in age of onset but are best understood within the context of neurodevelopment. Here, we review opportunities for intervention at critical points in developmental trajectories. We begin by discussing potential opportunities to prevent neuropsychiatric disorders. Once symptoms begin to emerge, a number of interventions have been studied either before a diagnosis can be made or shortly after diagnosis. Although some of these interventions are helpful, few are based upon an understanding of pathophysiology, and most ameliorate rather than resolve symptoms. As such, in the next portion of the review, we turn our discussion to genetic syndromes that are rare phenocopies of common diagnoses such as autism spectrum disorder or schizophrenia. Cellular or animal models of these syndromes point to specific regulatory or signaling pathways. As examples, findings from the mouse models of Fragile X and Rett syndromes point to potential treatments now being tested in randomized clinical trials. Paralleling oncology, we can hope that our treatments will move from nonspecific, like chemotherapies thrown at a wide range of tumor types, to specific, like the protein kinase inhibitors that target molecularly defined tumors. Some of these targeted treatments later show benefit for a broader, yet specific, array of cancers. We can hope that medications developed within rare neurodevelopmental syndromes will similarly help subgroups of patients with disruptions in overlapping signaling pathways. The insights gleaned from treatment development in rare phenocopy syndromes may also teach us how to test treatments based upon emerging common genetic or environmental risk factors.
Similar content being viewed by others

INTRODUCTION
Although neuropsychiatric disorders vary substantially in age of onset, ranging from toddlers with autism spectrum disorder to young adults with schizophrenia, it is increasingly apparent that these disorders are best understood within the broader context of neurodevelopment. Some disorders may be best described as manifestations of symptoms arising during the earliest points in development; others represent emerging deficits relative to typically developing peers over time; and still others present as rapid onset of new behavioral symptoms after a period of apparently intact development. Increasing data suggest that disorders traditionally categorized and described during adulthood, such as schizophrenia (see Schmidt and Mirnics, 2015, this issue), are associated with early vulnerabilities and deficits during childhood and adolescence.
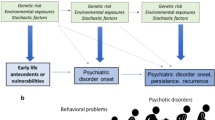
Recognizing the developmental context of neuropsychiatric disorders allows a movement toward more powerful treatment and prevention efforts. In theory, the earlier an intervention can be effectively deployed, the greater the long-term impact. The classic model, as shown in Figure 1, is that these disorders diverge gradually from health over time, such that intervention at earlier points may yield more powerful and long-lasting impact over the lifespan. This view posits that illness represents the outcome of a cascading process, as may be true for a child with behavioral inhibition at 3 years old who develops social phobia at 9 and depression at 20 (Beesdo et al, 2007; Lahat et al, 2014; Pine et al, 1998). As reviewed by Henderson et al, 2015, this issue, intervention with a subset of children with behavioral inhibition may improve symptoms at 3 and also prevent the amplification of illness across adolescence and young adulthood, thereby profoundly altering a child’s life course.

Neuropsychiatric disorders sometimes diverge gradually from health over time, such that intervention at earlier points may yield more powerful and long-lasting impact over the lifespan. In this model, eventual impairment is the result of cascading effects across development, as initial deficits prevent acquisition of skills later in development. Intervention early in the course of development may halt these cascading effects and thereby prevent the amplification of initial deficits.
One risk of beginning interventions during earlier developmental windows may be that adverse events related to treatment may also cascade over time. At the simplest level, early weight gain from atypical antipsychotic drugs may fundamentally change a child’s body habitus and risk of metabolic disorders for a lifetime (Maayan and Correll, 2011). As reviewed in this issue by Suri et al (2015), early exposure to psychotropic medications in animals can lead to long-term changes in behavior that extend well beyond the usual timeframe of treatment studies. We have a limited understanding of how these observations may extend to humans, but medication responses in children do clearly differ from adult responses. As one example, child psychiatrists sometimes use the term ‘activation’ to describe transient symptoms that resemble mania in children prescribed antidepressants (Goldsmith et al, 2011; King et al, 2009). It remains unclear whether this drug response is predictive or may even serve to kindle risk of later bipolar illness (Goldsmith et al, 2011).
The first section of our review will discuss the appealing opportunities for treatment at critical points in development, highlighting the current evidence for interventions as well as areas that are ripe for further progress. Although some of these treatments are clearly helpful, few are based upon an understanding of pathophysiology and most ameliorate rather than resolve symptoms. As such, the second section will turn to examples of specific genetically mediated neurodevelopmental disorders where model systems suggest treatments that could be transformative. Discussion of these targeted interventions will bridge to the third section, which will focus on challenges to understanding and deploying interventions in the context of neurodevelopment.
ARE BEHAVIORAL DISORDERS, SIMILAR TO CANCERS, MORE TREATABLE IF YOU CATCH THEM EARLY?
The Possibility of Prevention
Ideally, interventions would be applied based upon the earliest deviation from mental health, so that later impairment never manifests and symptoms never become a ‘disorder.’ Such preventive treatments could even be applied based not upon symptoms themselves but upon apparent risk factors that predict later symptoms. This hope of early actionable concerns has led investigators to push prevention research toward earlier and earlier points of development. In schizophrenia research, children or adolescents are treated at the point when familial risk and early symptoms converge (Miklowitz et al, 2014). In ASD research, high-risk populations such as younger siblings are tracked during infancy to identify the first warning signs of atypical trajectory that may in turn be treated incrementally from low- to high-intensity approaches as the specific trajectory and response of the child becomes clearer (Green et al, 2013). Before reasonably applying such treatments, we need to better understand the longitudinal time course of initial symptoms that predict onset of a disorder. As discussed below, we are closer in some areas than in others, and, unfortunately, in many circumstances, our work on early detection exceeds our capacity for treatment of these early concerns.
When evaluating prevention strategies, there are three main approaches. Primary prevention is applied across a population to prevent a risk factor from occurring in the first place. Secondary prevention is targeted at the point between occurrence of a risk factor and manifestation of symptoms that merit a diagnosis. Tertiary prevention is applied after symptoms have begun in order to ameliorate the severity of a disorder. This last form of ‘prevention’ blurs into early treatments that are applied at initial diagnosis. We will therefore include this strategy in our discussion of interventions below.
Primary prevention strategies are typically applied from a public health perspective, not on an individual level. With respect to neurodevelopmental disorders that include prominent cognitive symptoms such as ASD or schizophrenia, emerging data suggest that primary prevention should focus most logically on pregnancy, when the blueprint for the brain is laid out. Several examples of prenatal prevention strategies have emerged; although it is not always possible to know the full impact of population-level interventions. Despite societal concerns about vaccines (not supported by scientific evidence (Taylor et al, 2014)), the Measles–Mumps–Rubella vaccine has likely prevented many cases of ASD, since in utero exposure to rubella during the late first trimester is a robust risk factor for ASD (Chess, 1971). Similarly, the recommendation for pregnant women to not change cat litter has likely decreased risk by avoiding exposure to Toxoplasma gondii, a parasite that is a risk factor for both schizophrenia and suicidality (Pedersen et al, 2011, 2012). Recent data suggest that prenatal folate supplementation may serve to decrease risk of ASD, suggesting that widespread supplementation in the past few decades may have prevented some cases of ASD (Suren et al, 2013). As reviewed by Ross et al, 2015, this issue, in utero exposures to either drugs of abuse or certain prescription medications confer risk of cognitive and behavioral problems. Such prevention strategies to date are most appropriately understood from a risk perspective with very limited understanding of how this reduced risk translates into actual prevention of the neurodevelopmental disorders either on an individual or group level.
Fewer data point to straightforward primary prevention strategies that can be applied after birth. Initial brain wiring is very vulnerable to environmental exposures and is dependent upon the support of the placenta and the protective environment of the uterus. Later brain development may be less vulnerable to chemical, infectious, or immune exposures but is dependent upon experience, and either inadequate or damaging environments confer substantial long-term risk. Neglected children from Romanian orphanages manifest symptoms that are similar in some ways to autism spectrum disorder (Rutter et al, 1999). Much sociological work has focused on the long-term impact of ‘toxic stress’ in childhood (Shonkoff et al, 2012). Decreasing stress levels for families of young children, as well as preventing abuse and neglect, could have a huge impact on promoting later mental health. These goals, however, step into a political arena that often does not view them from a public health perspective.
Secondary prevention strategies depend upon knowing not just what factors confer a substantial risk of a neuropsychiatric disorder, but also how to prevent that risk from being realized. If the risk factor is less strong or the prevention strategy is less potent, then the number needed to treat to prevent one occurrence of the disorder will be too high to reasonably study. The classic example of secondary prevention of neurodevelopmental disorders is phenylketonuria (PKU), a genetic absence of phenylalanine hydroxylase, which universally causes intellectual disability, seizures, and frequently autism spectrum disorder in children fed a typical diet. Newborn screening for PKU is now universal in the United States and can identify PKU within the first few weeks of life, before neurodevelopmental symptoms are present. If an infant is found to have PKU, then a phenylalanine-free diet is implemented and often prevents lifelong disability. The number needed to treat in order to prevent disability in PKU is 1, as it universally leads to disability without intervention and the diet rescues most impairment in every child. This not only makes the intervention worthwhile, but it makes it easy to study, not even requiring a placebo-controlled trial. Further, developmental delay in PKU is evident before 2 years of age, allowing a much shorter duration of study than for most behavioral or cognitive impairments (Camp et al, 2014).
In contrast, if a risk factor only contributes a 10% increase in risk, as has been true for most common genetic variants identified in schizophrenia (Wright, 2014), then it becomes very difficult to demonstrate successful secondary prevention. Schizophrenia has a prevalence rate around 1.0%, so people with the risk factor would have a risk of schizophrenia of 1.1%. Even if we had a perfect preventative measure specific to that risk factor, we might need to treat 1000 people with the risk factor to prevent one case of schizophrenia. Studying such a preventative strategy is clearly impractical using conventional controlled research designs. Furthermore, the follow-up time for such a prevention trial may need to be decades in order to capture schizophrenia onset. Drawing a parallel with neurodegenerative disorders, the risk of Alzheimer’s disease (AD) is increased threefold by the common APOE ɛ4 risk allele, but it is very difficult to power and conduct a trial to assess prevention using medications that have been shown to be effective in slowing the rate of decline in AD. In contrast, secondary prevention studies in individuals with rare, highly penetrant risk genes for early onset AD, such as Presenilin-1 disruptions, are underway with sample sizes of only a few hundred people (Miller, 2012). Secondary prevention strategies in neuropsychiatry may therefore be limited, at least in the near-term, to individuals with very robust risk factors such as simple genetic disorders with high penetrance for neurodevelopmental symptoms (RR 10–100, see the discussion of Fragile X syndrome (FXS) below), those having an affected first-degree relative (RR 10–20 in the case of ASD or schizophrenia), or those having a severe medical risk factor, such as extreme preterm birth with very low birthweight (RR ∼10). Over time, we can hope that gene–gene or gene–environment interactions may be identified that will allow composite predictions of risk that approach the power of these individual risk factors, but that remains theoretical at this point (Iyegbe et al, 2014).
One robust risk factor for neuropsychiatric disorders is early neglect or abuse. Currently, in the United States, this neglect typically happens when a child is in the custody of a biological or foster family that is unable to provide adequate care. In other areas of the world, orphaned children may be placed instead into an orphanage setting, with various levels of contact with caregivers. As reviewed in detail by Humphreys and Zeanah (2015), this issue, a randomized study in Romania demonstrated that foster care leads to significantly better outcomes than remaining in the institutional setting (Nelson et al, 2007). For ethical and political reasons, similar studies are not available to assess the point at which it is better for a child to be placed in state foster care vs remaining with a biological family that is struggling to provide appropriate care. Further research is needed to better understand the impact of secondary prevention measures in the context of early parenting failure.
The Earlier the Better? Risk vs Impairment when Contemplating Intervention
Intervention strategies being pursued for early signs of schizophrenia, autism spectrum disorder, and mood disorders are very similar to tertiary prevention, with the goal to catch the very earliest symptoms and intervene before impairment is severe. The firmly held belief in the pediatric mental health community is that the earlier an intervention is started, the better the outcome is likely to be. This is a logical belief and matches the strong desire to help vulnerable children; although more work needs to be done to test the specific interventions and timing across different neuropsychiatric disorders.
The best examples of interventions that target symptoms pre-diagnosis are in high-risk youth presenting with initial symptoms in the months to years before meeting full criteria for schizophrenia or bipolar disorder. Investigators have used various strategies to enrich populations for risk of progressing to these diagnoses. Most studies have recruited adolescents at the point when they are first manifesting perceptual, mood, cognitive, or negative symptoms but do not yet meet severity or duration criteria for a diagnosis. Although this group is at much heightened risk, the majority do not go on to receive a diagnosis within 2 years. Some promising data have emerged for the use of cognitive-behavioral therapy, family therapy, atypical antipsychotic medications, and omega 3 fatty acids to prevent progression to schizophrenia, but these initial findings are quite mixed and await replication (Marshall and Rathbone, 2011; Miklowitz et al, 2014). One concerning finding is that while some could potentially be protected from progressing to a psychotic disorder, all of those who are treated are exposed to side effects of medications, most notably weight gain because of atypical antipsychotic medications including risperidone and olanzapine (Marshall and Rathbone, 2011). This is an area where clear data on the number needed to treat to prevent symptom progression will be very important for evaluating the cost vs benefit for vulnerable children.
The most promising example of early treatment having an impact on disease course is in autism spectrum disorder, where early, intensive behavioral intervention (EIBI) shows significant evidence for improving cognitive and early functional outcomes when applied in toddlers and preschool children with ASD. A number of variations of EIBI have been described, but all harness basic behavioral principles and administer intensive intervention programs (ie, >15 h per week) over substantial intervals of time (ie, 1–2 years). Typical improvements in intervention groups appear to be about 1 SD in IQ (15 points), with smaller but still significant improvements in adaptive behavior in most studies (Warren et al, 2011a). These studies have not been designed to assess whether the age of starting the intervention predicts outcome, but limited indirect data support this idea. The most notable comprehensive early intensive behavioral intervention study took children diagnosed with ASD before 24 months of age and observed an improvement in outcomes at 4 years, but few children moved off of the autism spectrum (Dawson et al, 2010). Even in this case, though, intervention was initiated after the child was impaired enough to receive an ASD diagnosis. From this perspective, intervention for many neuropsychiatric disorders does not yet often represent prevention of the disorder; although it may prevent many of the known secondary impairments associated with the disorder (eg, intellectual disability and language function in the case of ASD). Adult outcome studies are limited, but the available data suggest that early response to treatment, particularly treatment before 3 years of age, is associated with optimal cognitive functioning in adulthood (Anderson et al, 2014).
Pushing interventions to the age of first actionable concerns requires a clearer longitudinal picture of disorder onset within the context of numerous and varied developmental trajectories. In the case of ASD, younger siblings of affected children have a risk between 8 and 17% of developing the disorder themselves (Ozonoff et al, 2011). Multiple research groups have followed these ‘baby siblings’ over time to probe for early symptoms that may predict later diagnosis. To date, the observed early deficits, such as delayed postural control at 6–9 months or decreased eye gaze to social stimuli at 12 months, show limited predictive ability in this high-risk design. Recent data suggest that it is the trajectory of social development that indicates emerging symptoms, rather than deficits at a specific time point (Jones and Klin, 2013). This matches neuroimaging data that point to an altered trajectory of brain connectivity that is only evident longitudinally (Wolff et al, 2012). The cost of early intensive intervention combined with significant resource limitations make it daunting to recruit 6- to 10-fold greater sample sizes to evaluate secondary prevention approaches of comprehensive interventions in baby sibling samples. Perhaps, though, we are ready for tertiary prevention studies that target 9- or 12-month-old baby siblings who show early symptoms of social decrements or repetitive behavior. The high recurrence of other impairing communication and behavioral disorders beyond ASD (Messinger et al, 2013; Ozonoff et al, 2014) in this population may also suggest treatment approaches that begin by targeting broad risk and step up their intensity for those children who fail to follow a typical developmental trajectory, as has been piloted in studies with sequential, multiple assignment, randomized trial (SMART) designs (Kasari et al, 2014). Ultimately, moving beyond this high-risk group is more challenging as social and language development is quite variable in toddlers, making it difficult to differentiate between low-risk children who are delayed but will catch up on their own versus children who are showing symptoms that will develop into later functional impairments.
The Potential for Arresting Cascades of Emerging Symptoms Across Neurodevelopment
Some early developmental differences may serve as pivotal areas of vulnerability that subsequently result in later impairment. For example, a 9-month-old boy may display an exceptional preference for certain specific objects relative to developmentally typical interest in social stimuli, resulting in a less socially rich environment over time. Consequently, he may be less likely to attend to speech or coordinate his attention with others, and therefore be slower to develop later skills, including speech. In this example, early deficits in social attention shift the environmental inputs needed for optimal neurodevelopment and social learning. Early neurodevelopmental symptoms may compound over time by affecting pivotal learning processes and the complex, transactional interplay between the boy and his caregiver. In turn, an intervention targeted to these early deficits could impact not just his specific social communication and attention deficits but also his ability to acquire many other skills normally absorbed in the context of social interaction over time. Again this theoretical approach is quite appealing, but to date the intervention literature has yet to specifically pinpoint these early pivotal skills and demonstrate that measured changes in these specific areas mediate the posited neurodevelopmental cascade, as shown in Figure 1.
A few intervention approaches may be particularly promising for cascading benefits. One example may be interventions at the level of the family, who may then be able to better care for a child over the course of his or her development. Miklowitz et al (2013) describe more rapid recovery from mood symptoms and more weeks without mood symptoms in children at familial risk of bipolar disorder randomized to family-focused psychotherapy in comparison with a control group. Early treatment approaches in ASD often include parent training that promotes use of new skills in the home setting. Although training parents is more cost effective than using only clinician-administered treatment, findings using parent-based interventions have been mixed thus far in ASD intervention (Rogers et al, 2012).
Individual or group psychotherapy approaches could also lead to cascading benefit by promoting those behaviors or thoughts that foster mental health. For example, helping children focus on feelings of pleasure or accomplishment could become self-reinforcing over time. Garber et al (2009) conducted a large randomized trial of group cognitive-behavioral therapy in adolescents who had a parent with a past or current diagnosis of depression. These adolescents, who themselves had past diagnoses of depression or current sub-threshold depression symptoms, showed a lower rate of depression diagnosis in the 3 years subsequent to intervention (Beardslee et al, 2013), but this benefit was not seen if a parent was depressed at the point of study entry. Other psychotherapy approaches, such as attention bias modification treatment, described in this issue by Henderson et al (2015), explicitly target a child’s focus on stimuli in their environment in real time (Shechner et al, 2014).
If interventions early in the disease course do indeed arrest neurodevelopmental cascades that lead to worsening impairment over time, then even relatively intensive treatments may be cost effective when viewed across the lifespan. Again, this is most stark with the most severe disorders. In the case of ASD, estimates place the lifetime cost of caring for a single individual at two to three million dollars (Ganz, 2007; Knapp et al, 2009). EIBIs for ASD frequently require 15+ hours of therapy each week and may be delivered for several years, but the yield of an average IQ improvement of 15 points may still provide a tremendous return on investment on an individual, family and system level. In other conditions, such as childhood cancer or birth complications, we spare no expense to improve length or quality of life. In contrast, insurance companies have resisted covering expensive behavioral interventions, with family advocacy and legislation often being necessary to force coverage. A longitudinal perspective suggests that intervention early in the disease course may be the most cost-effective approach for most neurodevelopmental and mental health diagnoses (Motiwala et al, 2006); although firm data on cost-effectiveness remain sparse.
TARGETED TREATMENT AS THE NEXT STEP FOR TREATMENTS DURING DEVELOPMENT
Common Risk Factors May Point to Neurobiological Systems for Further Study
Most medication treatments for neuropsychiatric disorders were discovered by happenstance coupled with careful clinical observation, rather than emerging from an understanding of pathophysiology. With respect to typically categorized childhood-onset disorders, the stimulant medication benzedrine was first found to benefit attention and school performance when given to children in the 1930s to attempt to alleviate headaches after lumbar puncture (Bradley, 1937; Strohl, 2011). The first medications in most major psychotropic classes were similarly discovered after being developed or used for another purpose, including lithium (gout), chlorpromazine (antihistamine), and monoamine oxidase inhibitors (anti-tuberculosis) (Lopez-Munoz and Alamo, 2009; Shen, 1999). The mechanism of action for these medications was often discovered well after their therapeutic efficacy was described, and, in most cases, we still do not fully understand the mechanisms of action.
Environmental and genetic risk factors have been identified for many neurodevelopmental disorders, leading us closer to an understanding of pathophysiology. Most of these factors, however, contribute a relatively modest amount of risk. For example, epidemiological studies suggest that cannabis use during adolescence is associated with an approximately two- to three-fold increase in later diagnosis of schizophrenia, with earlier exposure associated with greater risk (Casadio et al, 2011); although the mechanism is unclear. This may be an important component of risk, but you would expect only a very small number of marijuana users to develop schizophrenia. Within that number, you may expect to see a necessary convergence with other risk factors, including genetic variants (Caspi et al, 2005). On the genetic front, the strongest common polymorphism finding in a recent large meta-analysis of whole-genome association data in schizophrenia had an odds ratio of 1.2 (Ripke et al, 2013), suggesting that many different polymorphisms—and likely environmental risk factors as well—would have to converge to lead to substantial risk.
On the positive side, it is possible that a common risk factor with relatively small effect size could correspond to a neurotransmitter or signaling pathway that could generate a novel, high-impact treatment. As a hopeful example, a recent meta-analysis of genome-wide association data found that the polymorphisms near the preproinsulin precursor gene INS have an odds ratio of <1.1 in type 2 diabetes (Saxena et al, 2012). Despite this small impact on risk, the downstream gene product, insulin, remains the definitive treatment for many people with diabetes. A parallel example may be the modest but statistically significant association of the dopamine receptor D2 gene DRD2 with schizophrenia (Wright, 2014). Even while we wish for better treatments, medications that target the D2 receptor remain our most powerful option in schizophrenia (Masri et al, 2008).
Unlike targeted treatment development, however, insulin and antipsychotic medications were first identified as treatments and later supported by the identification of genetic risk loci. It may be more challenging to work forward to new treatments from genetic variants that make small contributions to risk. Animal models of such risk factors would not be expected to show behavioral phenotypes that precisely parallel the human condition. We could hope, however, that studying the implicated systems could lead to insights into the underlying neurobiology, even if an animal model does not directly mimic the disorder itself. As one example, human genetic studies implicate variation in the serotonin transporter gene promoter in susceptibility to depression in the face of childhood maltreatment (Karg et al, 2011). Humans lacking the serotonin transporter entirely have not been identified, but the serotonin transporter knockout mouse has taught us much about the importance of serotonin clearance across development (Murphy and Lesch, 2008), which we can hope would point to new mechanisms for treatment.
Rare, Simple Genetic Phenocopies Could Reveal New Treatment Pathways
Individuals with relatively simple genetic syndromes are frequently found within large, heterogeneous groups of people with neuropsychiatric diagnoses such as schizophrenia or ASD. These rare syndromes often show up across multiple diagnostic categories, suggesting risk of neurobehavioral impairment that is modified by other developmental, environmental, or genetic factors. For example, deletion of chromosome 22q11.2 results in velocardiofacial syndrome (VCFS), which is associated with diagnosis of intellectual disability, ADHD, and ASD in childhood (Hooper et al, 2013), mood disorders in adolescence (Jolin et al, 2012), and psychosis in adulthood (Green et al, 2009). As an even rarer example, a balanced translocation in one family identified disruption of the DISC1 gene as a highly penetrant risk factor for schizophrenia and mood disorders (Millar et al, 2000).
Most data point to common genetic and environmental risk factors as contributing risk in the majority of people with a given neurodevelopmental disorder, so why should we focus on rare phenocopy syndromes? Unlike common gene variants that frequently result from relatively subtle changes in gene expression, rare genetic phenocopy syndromes typically result from a complete disruption (or duplication) of a gene or chromosomal region, either on one or both chromosomes. Such a disruption can be easily modeled in cellular and animal models. Further, such models can be expected to yield neurobiological insights when the rare phenocopy shows an odds ratio of 30–60 for a behavioral phenotype, in contrast to the typical common risk factor yielding an odds ratio of 1.1–2. With the emergence of new genome-editing technology, models of gene disruption can be produced in a few weeks or months (Burgess, 2013). Mouse models of some of these relatively simple genetic syndromes, including both VCFS and DISC1 disruption, are beginning to yield an understanding of the neurobiology of these rare phenocopies of neurodevelopmental disorders. Although these syndromes are individually rare, they collectively represent a substantial minority of cases, and their relative simplicity may provide a clearer path to developing treatments that may benefit a larger group of individuals who share both neuropsychiatric symptoms and underlying perturbations in neurodevelopment.
FXS is the rare neurodevelopmental phenocopy syndrome that has progressed the furthest toward an understanding of pathophysiology. FXS is an X-linked recessive disorder that is the most common inherited cause of both intellectual disability and ASD, accounting for approximately 1–2% of affected individuals (Hagerman et al, 2009; Loesch et al, 2007). An expanding trinucleotide repeat in the 5′ untranslated region of the FMR1 gene causes FXS (Kremer et al, 1991). Genetic testing for FXS is now recommended for all children with intellectual disability or ASD. Children with FXS frequently show hyperactivity, anxiety, sensory sensitivity, and avoidance of eye contact (Hagerman et al, 2009). Approximately 30–60% of individuals with FXS meet criteria for ASD; although patterns of symptoms differ from the general ASD population (Hall et al, 2010; Harris et al, 2008).
Much progress has been made in understanding the molecular changes that connect loss of FMR1 expression to observed cognitive and behavioral symptoms (Heulens and Kooy, 2011). Mice lacking Fmr1 actually show fairly subtle behavioral deficits, including mild hyperactivity, subtle learning deficits, and decreased initial exploration of a novel animal. In contrast, their neuropathology closely parallels human FXS, with increased numbers and length of dendritic spines, corresponding to an immature appearance. As one explanation for failure to form mature synapses, Huber et al (2002) first identified changes in long-term depression, a form of synaptic plasticity, in Fmr1 knockout mice. These studies led Bear et al (2004) to propose the ‘mGluR theory’ of FXS. They posited that, in the absence of the Fragile X protein, which binds to the mRNA of multiple post-synaptic proteins, group 1 mGluR stimulation results in excessive trafficking of AMPA receptors out of the recirculating pool and enhanced long-term depression. They tested this hypothesis in Fmr1 knockout mice by reducing by half the normal expression of mGlu5 receptor (Dolen et al, 2007), demonstrating a rescue of protein synthesis, dendritic spine, and behavioral phenotypes. A number of studies have now reported rescue of multiple phenotypes in Fmr1 knockout animals with mGlu5 receptor-negative allosteric modulator drugs with different selectivity at various points in development and with a range of treatment duration (Michalon et al, 2012).
Although the mGlu5 theory of FXS has the most support from genetic and pharmacological rescue studies in mice, a number of other potential treatments have also been proposed, including GABA receptor agonists, minocycline, and lithium (Bagni and Oostra, 2013; Gross et al, 2012; Henderson et al, 2012; King and Jope, 2013). With multiple pharmacological approaches rescuing some brain or behavioral phenotypes in Fmr1 knockout animals, the overall data suggest either multiple pathways toward treatment or a lack of specificity to some of these rescued phenotypes. Testing these various potential treatments in humans may help us understand the indications that an approach will—or will not—translate across species.
A similar story of potential treatment is emerging in Rett syndrome, where X-linked dominant loss of MECP2 gene function leads to regression in cognitive development, speech, and motor function, typically in the second year of life (Amir et al, 1999). Motor function continues to worsen through the second decade of life, when breathing abnormalities emerge and are the most common cause of death. The encoded protein, methyl CpG binding protein 2, regulates expression of multiple genes, including BDNF, encoding brain-derived neurotrophic factor. Returning Mecp2 gene function in adulthood rescues synaptic function as well as motor decline and premature death observed in the mouse model of Rett syndrome, suggesting that treatment may be possible even after the period of initial regression (Cobb et al, 2010).
In addition to multiple experiments showing rescue with Mecp2 itself, overexpression of the BDNF gene leads to improvements in synaptic function and motor impairment, as well as prolonged survival in Mecp2 knockout animals (Chang et al, 2006). BDNF itself is not practical as a therapy because it does not cross the blood–brain barrier. Most hypotheses about potential treatments therefore focus on drugs that may increase BDNF signaling, such as insulin-like growth factor 1, which improves synaptic function, motor impairment, and early death in mice lacking Mecp2 (Tropea et al, 2009). Treatment with fingolimod, a drug that increases BDNF levels, has also been reported to improve motor function and brain region shrinkage in these animals (Deogracias et al, 2012). Treatment with an agonist for the BDNF receptor, trk-B, leads to improvement in apnea and biochemical markers of BDNF signaling in mice with decreased Mecp2 expression (Schmid et al, 2012).
A number of other phenocopy syndromes have enough data to suggest pathways for treatment in humans. Rapamycin rescues brain and behavioral phenotypes in a mouse model of tuberous sclerosis complex, which frequently includes intellectual disability and autism spectrum disorder (Ehninger et al, 2008). Low-dose clonazepam, a GABA-A receptor-positive allosteric modulator, rescues social and cognitive deficits in a mouse model of Dravet syndrome, an epilepsy and intellectual disability syndrome also sometimes associated with ASD (Han et al, 2012). Lithium rescues vesicle transport in a cellular model of Disc1 deletion (Flores et al, 2011). Each of these rare genetic phenocopy syndromes may reveal new pathways toward treatment, with some of these pathways possibly extending to a broader group of children with similar behavioral symptoms.
CHALLENGES TO STUDYING TREATMENTS IN THE CONTEXT OF DEVELOPMENT
Utility of Rodent Models: Mice Are not Little Humans
Rodent models have been critical to understanding the underlying pathophysiology of many human diseases. As noted above, rodent models of simple genetic phenocopies of neurodevelopmental disorders offer particularly potent opportunities to dissect the downstream consequences of robust risk factors of disease. The remarkable discoveries in mice lacking Fmr1 and Mecp2 offer hope for conceptualizing treatments in disorders that most assumed would be untreatable past the earliest years of childhood (van der Worp et al, 2010). With the enormous differences between rodents and humans, however, we must embrace the fact that translating treatments between species will not be simple.
It goes without saying that the mouse brain is much, much smaller than the human brain. Furthermore, the cortex occupies a much smaller fraction of the total brain in the mouse, and it lacks the convolutions that increase cortical surface area in the primate. Comparative studies reveal many differences in cortical gene expression in the brain of mice vs human. As one example particularly relevant to neurodevelopmental disorders, FMR1 shows a marked difference in developmental cortical expression between rodents and primates. The rich expression of FMR1 in developing human cortex appears to regulate expression of nitric oxide synthase in an alternating pattern across adjacent minicolumns, the vertical units of connection that span cortical layers (Kwan et al, 2012). This pattern of expression is completely absent in the mouse, calling into question the degree to which late rescue of behavioral and neuronal function in the Fmr1 null mouse will translate to fully developed humans with FXS.
Beyond differences in brain development and architecture, cognition and behavior differ quite substantially across species. Again, this is fairly obvious, as humans keep mice in small cages and manipulate their gene expression and environmental exposures, and not vice versa. It remains important to emphasize that the human brain coordinates extremely complex social, communicative, and behavioral processes that are only modeled on basic levels in animals. As the most complex observable behavior in humans, abnormalities in social behavior are particularly difficult to model in rodents, in part because they have minimal observable vocal communication. The dominant social paradigm in mice focuses on time spent exploring a novel mouse (Silverman et al, 2010), which is quite different than our complex assessment of social behavior in a human child (Lord et al, 2000). Without verbal communication, investigators define ‘schizophrenia-like’ behavior in mice using surrogate measures like prepulse inhibition or response to a psychotomimetic drug (Siuta et al, 2010). From a cognitive perspective, mice would perform at the floor (or sub-floor) of human intelligence testing. Despite many attempts, behavioral neuroscientists identify minimal evidence of cognitive impairment in Fmr1 null mice, which models the most common inherited cause of intellectual disability. Genetic and drug rescue of brain and behavioral phenotypes in these animals most clearly impacts hyperactivity and seizure susceptibility, making it difficult to pick appropriate cognitive and behavioral targets for human studies (Michalon et al, 2012).
The short reproductive cycle and rapid development of lower mammal species are major advantages for conducting research studies, but they also offer challenges to translation. Most rodent species are born at the equivalent of human third trimester. The defined critical period for exposure to serotonin reuptake inhibitors leading to adult anxiety-like behavior is immediately postnatal in the mouse, which would largely correspond to the third trimester in the human (see Suri et al, 2015, this issue). Mouse pups open their eyes at 1 week and transition to solid food at 3 weeks, when they are a mere 3–5 weeks from sexual maturity. The best study of an mGlu5 receptor negative allosteric modulator in Fmr1 null animals administered treatment for 4 weeks: from shortly after weaning all the way through to sexual maturity (Michalon et al, 2012). This would correspond to a decade of treatment in the human. The Mecp2 null mouse shows a period of typical motor development followed by a regression that is quite similar to the human pattern, but the regression happens in adult mice instead of 6–18-month-old girls with Rett syndrome (Guy et al, 2001). In contrast, studies of serotonin reuptake inhibitors in mouse and human reveal effects of chronic treatment emerging only after 3–6 weeks, suggesting similar patterns of receptor accommodation over time across species (Dulawa et al, 2004). The uncertainty of matching windows of mouse development to human development raises major questions for investigators seeking to translate across species. For example, should we match duration of treatment (eg, 4 weeks) or developmental window of treatment (eg, 10 years)? The correct answer is likely a compromise that emphasizes an early treatment window with enough duration to anticipate meaningful functional improvement.
Ethical and Regulatory Implications for Early Treatment: Children Are not Little Adults
Given the uncertainties in parallels between rodent and human brain development, it seems reasonable to assume that treatment benefits for neurodevelopmental disorders will be most robust either before or soon after the emergence of symptoms. This will often mean conducting initial treatment studies in children or adolescents, which raises ethical and regulatory issues, as well as measurement challenges (see subsection below). From a regulatory standpoint, phase I safety and dose-finding (pharmacokinetic) studies are conducted in consenting adults. After phase I, investigators and regulators are presented with some dilemmas about moving studies into the patient population. (1) Should safety and dose-finding studies be conducted in an adult population manifesting the later impairments of the disorder before moving down to affected adolescents and then to children who may be evidencing differing vulnerabilities within very different neurodevelopmental periods? (2) Does the priority given to testing medications first in those with the ability to consent (ie, adults) hold when the affected adult population has decreased capacity to assent and requires a surrogate decision-maker? (3) Should we look for efficacy along with safety and dose finding in adults with neurodevelopmental disorders, or should we focus our studies of efficacy primarily on the younger age when benefit is most likely to be seen? (4) Given the hypotheses and theoretical framework positing substantial beneficial effects at very early points in development and much more limited impact at later times, how are we to safely and ethically translate therapeutics to early childhood, toddlerhood, or infancy?
The answers to these questions are by no means obvious from an ethical or practical perspective. Deciding to protect children by looking first for efficacy in adults may mean that a potentially efficacious treatment is abandoned because of lack of effect treatment signal before being tested in the population most likely to benefit. As potential examples, two drug development programs in FXS (Novartis NCT01253629 and NCT01357239, Seaside Therapeutics NCT01282268) were recently closed without testing efficacy in children below the age of 12 because efficacy studies in adults and adolescents showed no measurable benefit. It is possible that benefit would only be seen in early childhood, when cognitive development follows the steepest slope. On the other hand, exposing young children to a medication with an uncertain safety profile could result in a life-long change in developmental trajectory for the worse.
In contrast to disorders where onset by definition occurs in the context of neurodevelopment, such as ASD or ADHD, some disorders may have onset in children or adults with very similar patterns of symptoms that may or may not respond similarly to treatment. The available examples suggest that it is logical in such situations to test treatments first in adults and then move down into children. In the case of anxiety disorders and obsessive–compulsive disorder, similar benefits, side effects, and placebo response rates are seen in studies of serotonin reuptake inhibitors in adults and in children (Pediatric OCD Treatment Study (POTS) Team, 2004; Walkup et al, 2008). In the case of major depressive disorder, however, children and adolescent trials reveal greater placebo response and more risk of treatment-emergent suicidality with serotonin reuptake inhibitors in contrast with adult trials (Hammad et al, 2006; Tsapakis et al, 2008). Similarly, efficacy data are mixed in trials of medication for pediatric bipolar disorder (Liu et al, 2011), with some evidence for more weight gain in the younger population, which could lead to a lifelong change in metabolic profile (Correll et al, 2010). Overall, the contrast between treatment results in children and adults highlights the importance of careful studies extending into children before drawing conclusions about potential efficacy or harms in disorders that manifest across development.
Measurement Challenges: Differentiating Treatment Effects from Ongoing Development
Designing and testing interventions that impact developmental trajectories at the age of earliest concern is a critical research priority, but it has been difficult to achieve consensus on how change in response to such interventions should be measured. Many different outcome measures have been applied across studies, even within single diagnostic entities (Warren et al, 2011b). The challenges extend beyond establishing consensus measures to fundamental challenges in studying developing humans. First, even when they have neurodevelopmental disorders, children and adolescents (and young adults) continue to follow a developmental trajectory. This complicates measurement of treatment effects when parents or clinicians are asked to rate measures of cognitive, social, or communication function that can be expected to show some change across development. When continued development is matched by expectancy of improvement with treatment, we should not be surprised by the 30–50% placebo response rates seen for core symptom measures in ASD, for example (Sandler and Bodfish, 2000). The pace of cognitive development slows over time, so that treatment studies in older children or adolescents may have more ability to differentiate treatment response from developmental gain. Longitudinal studies in targeted populations, such as rare phenocopy syndromes, would help us better plan treatment studies by providing a better picture of the development of cognition and adaptive behavior over time.
A second challenge for outcome measures applied to children is that parents or guardians often must be the primary informants. The layering of informants means that the participants’ internal experience is filtered through the eyes of another, with variability in the parents’ experience and mood potentially amplifying variability in the participant. Further, the parent often observes the child for a minority of the day, and teacher ratings, while very useful, are much more difficult to obtain. Coupling parent report with objective measures of child symptoms, such as video coding or eye tracking, could give us a clearer sense of parents’ ability to accurately rate child behavior. As one example of the need for these comparisons, child ratings of anxiety correlated with cortisol response but parent ratings did not in one study of high-functioning boys with ASD (Bitsika et al, 2014).
A third challenge has been the wide variety of outcome measures used across treatment studies. This heterogeneity can be expected in the early phases of treatment research but should be followed by consolidation when one or a few outcome measures are shown to accurately index change in a particular symptom domain. This can be seen in ADHD research, where the most commonly used set of outcome measures can be shown to be both valid and reliable across time and treatment, particularly when used as a composite across measures and informants (Conners et al, 2001). In other areas, such as ASD or rare phenocopy syndromes, where there are fewer treatment studies with significant findings (or none), it is more difficult to identify a narrow set of outcome measures that will reliably detect change in symptoms. In such cases, the field must start by identifying consensus measures, which are likely to be imperfect, while waiting for more data to emerge (Anagnostou et al, 2014 (in press); Scahill et al, 2013). Studies that choose a less commonly used primary outcome measure should still include 1–2 standard measures to allow cross-measure comparison.
If subjective ratings by parents or clinicians are unreliable outcome measures because of ongoing cognitive development, expectancy bias, and expectable fluctuations in symptoms over time, objective measures may provide an alternative that allows clear measurement of symptom change. This is supported by the example of IQ as the most consistent indicator of change in clinical trials of EIBIs in ASD. One potential concern, however, is that some improvements may reflect behavioral training that improves cooperation with cognitive testing in the laboratory setting, rather than general improvements in cognitive function. Further, such changes may not be reflective of changes in core symptoms of the disorder itself. Checklists of adaptive behaviors that can be anchored to specific activities (eg, getting dressed independently, calling a classmate on the phone) also show differences between active treatment and the comparator treatment in some studies but are less consistent (Warren et al, 2011a). Other promising objective measures include fine-tuned observation such as video recording of behavior (Corbett et al, 2014; Stronach and Wetherby, 2014), audio recording of verbal exchanges over the course of a typical day (Warlaumont et al, 2014), performance in computer games (Andari et al, 2010), or eye gaze tracking (Jones and Klin, 2013). Proposed biomarkers of treatment response include assessment of brain response such as EEG (Dawson et al, 2012) or MRI (Gordon et al, 2013). Cross-validation studies of objective behavioral observations, biomarkers, and self-, teacher-, or parent-report questionnaires are critical to identify appropriate outcome measures for clinical trials.
Coupled with better, more objective measures of neuropsychiatric symptoms in developing humans, we need better approaches to evaluate moderators of treatment response. The heterogeneity of neurodevelopmental disorders like ASD or ADHD predicts that subsets of children are likely to respond to a given intervention, but we typically design studies to evaluate treatment effects in the overall group. As ongoing development and placebo or nonspecific effects of treatment account for the majority of improvement in most clinical trials, clinical trials may need to be structured to identify moderators of response to the active treatment in contrast with the placebo or another control condition. One recent example suggests that this approach may be especially well suited to disorders with considerable heterogeneity in symptom presentation. In contrast to a treatment as usual control group, Carter et al (2011) found no overall benefit for a parent-implemented intervention, Hanen’s ‘More than Words,’ on communication in children with ASD. A moderator analysis revealed, however, that children with less focus on objects at baseline showed significant gains, whereas children with more object focus actually worsened in comparison with the control group (Carter et al, 2011).
Moving past understanding what symptoms change and for whom, we need a better understanding of how short-term treatments relate to long-term outcomes. A 15-point improvement in IQ is impressive, but we do not have a clear understanding of whether such a change results in better educational attainment or long-term improvements in quality of life. In the Multimodal Treatment of ADHD (MTA) study, assignment to short-term stimulant treatment clearly showed a substantial impact on symptoms that separates easily from placebo. Unfortunately, outcomes measured 6–8 years after MTA study completion were not predicted by treatment in the randomized trial but were instead predicted by initial symptom trajectory, regardless of treatment (Molina et al, 2009). Longitudinal follow-up of promising short-term treatment studies, such as the recent Prevention Trial of Family-Focused Treatment in Youth at Risk for Psychosis (Miklowitz et al, 2014), is critical to understand the relationship between initial symptomatic response and meaningful long-term outcomes.
ALTHOUGH THE WAYS FORWARD WILL NOT BE EASY, SOME PATHS WILL LEAD TO NEW TREATMENTS
The complexity of human brain development may seem daunting, but our current position is not that different than the field of oncology a couple of decades ago. The FMR1 gene was identified in FXS about 20 years before the first clinical trials were initiated, a similar time window to cancer therapeutics (Fischgrabe and Wulfing, 2008; Xie et al, 2013). Brain development is clearly more complicated than tumor development, and we can expect that testing treatments for neurodevelopmental disorders will be more complicated as well. Although we can not simply measure tumor shrinkage or track mortality rates, we can carefully evaluate brain and behavioral parallels across species, rather than expecting global improvement in a neuropsychiatric disorder as a whole (Insel et al, 2010). We can carefully observe successful and unsuccessful trials to refine our methodology and outcome measures for the next set of studies.
As in oncology, we can expect that our initial treatments will be nonspecific, like the general chemotherapeutic agents thrown at a wide range of tumor types. Serotonin reuptake inhibitors, stimulants, and atypical antipsychotics may be the neuropsychopharmacology parallels of these broad-based agents. We should be glad to have these options, which help many children, but we should not be satisfied with them. With increasing understanding of neurodevelopment and of pathophysiology, we can expect to see more specific treatments emerge. These initial treatments may parallel imatinib (Gleevec), which was developed to treat a molecularly defined subset of chronic myelogenous leukemia but has subsequently been used to treat multiple other cancers (Zhang et al, 2009). We can hope that medications based upon the pathophysiology of Fragile X, Rett, or VCFS will similarly provide benefit for defined subgroups of patients with disruptions in overlapping regulatory or signaling pathways. The insights gleaned from treatment development in these rare phenocopy syndromes may also point the way to the right approaches, methodologies, and paradigms to test treatments based upon emerging common genetic or environmental risk factors.
FUNDING AND DISCLOSURE
Dr Veenstra-VanderWeele has consulted with Novartis, Roche Pharmaceuticals, and SynapDx, and has received research funding from Novartis, Roche Pharmaceuticals, SynapDx, Seaside Therapeutics, Forest, and Sunovion. Dr Warren has consulted with and received research funding from SynapDx.
References
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188.
Anagnostou E, Jones N, Huerta M, Halladay AK, Wang P, Scahill L et al (2014). Measuring social communication behaviors as a treatment endpoint in individuals with autism spectrum disorder. Autism (in press).
Andari E, Duhamel JR, Zalla T, Herbrecht E, Leboyer M, Sirigu A (2010). Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci USA 107: 4389–4394.
Anderson DK, Liang JW, Lord C (2014). Predicting young adult outcome among more and less cognitively able individuals with autism spectrum disorders. J Child Psychol Psychiatry 55: 485–494.
Bagni C, Oostra BA (2013). Fragile X syndrome: from protein function to therapy. Am J Med Genet A 161A: 2809–2821.
Bear MF, Huber KM, Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci 27: 370–377.
Beardslee WR, Brent DA, Weersing VR, Clarke GN, Porta G, Hollon SD et al (2013). Prevention of depression in at-risk adolescents: longer-term effects. JAMA Psychiatry 70: 1161–1170.
Beesdo K, Bittner A, Pine DS, Stein MB, Hofler M, Lieb R et al (2007). Incidence of social anxiety disorder and the consistent risk for secondary depression in the first three decades of life. Arch Gen Psychiatry 64: 903–912.
Bitsika V, Sharpley CF, Sweeney JA, McFarlane JR (2014). HPA and SAM axis responses as correlates of self- vs parental ratings of anxiety in boys with an autistic disorder. Physiol Behav 127: 1–7.
Bradley C (1937). The behavior of children receiving Benzedrine. Am J Psychiatry 94: 577–585.
Burgess DJ (2013). Technology: a CRISPR genome-editing tool. Nat Rev Genet 14: 80.
Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N et al (2014). Phenylketonuria scientific review conference: state of the science and future research needs. Mol Genet Metab 112: 87–122.
Carter AS, Messinger DS, Stone WL, Celimli S, Nahmias AS, Yoder P (2011). A randomized controlled trial of Hanen's 'More Than Words' in toddlers with early autism symptoms. J Child Psychol Psychiatry 52: 741–752.
Casadio P, Fernandes C, Murray RM, Di Forti M (2011). Cannabis use in young people: the risk for schizophrenia. Neurosci Biobehav Rev 35: 1779–1787.
Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H et al (2005). Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry 57: 1117–1127.
Chang Q, Khare G, Dani V, Nelson S, Jaenisch R (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49: 341–348.
Chess S (1971). Autism in children with congenital rubella. J Autism Childhood Schizophrenia 1: 33–47.
Cobb S, Guy J, Bird A (2010). Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans 38: 498–506.
Conners CK, Epstein JN, March JS, Angold A, Wells KC, Klaric J et al (2001). Multimodal treatment of ADHD in the MTA: an alternative outcome analysis. J Am Acad Child Adolesc Psychiatry 40: 159–167.
Corbett BA, Swain DM, Newsom C, Wang L, Song Y, Edgerton D (2014). Biobehavioral profiles of arousal and social motivation in autism spectrum disorders. J Child Psychol Psychiatry 55: 924–934.
Correll CU, Sheridan EM, DelBello MP (2010). Antipsychotic and mood stabilizer efficacy and tolerability in pediatric and adult patients with bipolar I mania: a comparative analysis of acute, randomized, placebo-controlled trials. Bipolar Disorders 12: 116–141.
Dawson G, Jones EJ, Merkle K, Venema K, Lowy R, Faja S et al (2012). Early behavioral intervention is associated with normalized brain activity in young children with autism. J Am Acad Child Adolesc Psychiatry 51: 1150–1159.
Dawson G, Rogers S, Munson J, Smith M, Winter J, Greenson J et al (2010). Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model. Pediatrics 125: e17–e23.
Deogracias R, Yazdani M, Dekkers MP, Guy J, Ionescu MC, Vogt KE et al (2012). Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci USA 109: 14230–14235.
Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S et al (2007). Correction of fragile X syndrome in mice. Neuron 56: 955–962.
Dulawa SC, Holick KA, Gundersen B, Hen R (2004). Effects of chronic fluoxetine in animal models of anxiety and depression. Neuropsychopharmacology 29: 1321–1330.
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ et al (2008). Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 14: 843–848.
Fischgrabe J, Wulfing P (2008). Targeted therapies in breast cancer: established drugs and recent developments. Curr Clin Pharmacol 3: 85–98.
Flores R 3rd, Hirota Y, Armstrong B, Sawa A, Tomoda T (2011). DISC1 regulates synaptic vesicle transport via a lithium-sensitive pathway. Neurosci Res 71: 71–77.
Ganz ML (2007). The lifetime distribution of the incremental societal costs of autism. Arch Pediatr Adolesc Med 161: 343–349.
Garber J, Clarke GN, Weersing VR, Beardslee WR, Brent DA, Gladstone TR et al (2009). Prevention of depression in at-risk adolescents: a randomized controlled trial. JAMA 301: 2215–2224.
Goldsmith M, Singh M, Chang K (2011). Antidepressants and psychostimulants in pediatric populations: is there an association with mania? Paediatric Drugs 13: 225–243.
Gordon I, Vander Wyk BC, Bennett RH, Cordeaux C, Lucas MV, Eilbott JA et al (2013). Oxytocin enhances brain function in children with autism. Proc Natl Acad Sci USA 110: 20953–20958.
Green J, Wan MW, Guiraud J, Holsgrove S, McNally J, Slonims V et al (2013). Intervention for infants at risk of developing autism: a case series. J Autism Dev Disord 43: 2502–2514.
Green T, Gothelf D, Glaser B, Debbane M, Frisch A, Kotler M et al (2009). Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J Am Acad Child Adolesc Psychiatry 48: 1060–1068.
Gross C, Berry-Kravis EM, Bassell GJ (2012). Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology 37: 178–195.
Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27: 322–326.
Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A et al (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123: 378–390.
Hall SS, Lightbody AA, Hirt M, Rezvani A, Reiss AL (2010). Autism in fragile X syndrome: a category mistake? J Am Acad Child Adolesc Psychiatry 49: 921–933.
Hammad TA, Laughren T, Racoosin J (2006). Suicidality in pediatric patients treated with antidepressant drugs. Arch Gen Psychiatry 63: 332–339.
Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB et al (2012). Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489: 385–390.
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I et al (2008). Autism profiles of males with fragile X syndrome. Am J Ment Retard 113: 427–438.
Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR et al (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABA(B) receptors with arbaclofen. Sci Transl Med 4: 152ra128.
Henderson HA, Pine DS, Fox NA (2015). Behavioral inhibition and developmental risk: a dual-processing perspective. Neuropsychopharmacology (in press).
Heulens I, Kooy F (2011). Fragile X syndrome: from gene discovery to therapy. Front Biosci 16: 1211–1232.
Hooper SR, Curtiss K, Schoch K, Keshavan MS, Allen A, Shashi V (2013). A longitudinal examination of the psychoeducational, neurocognitive, and psychiatric functioning in children with 22q11.2 deletion syndrome. Res Dev Disabil 34: 1758–1769.
Huber KM, Gallagher SM, Warren ST, Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99: 7746–7750.
Humphreys KL, Zeanah CH (2015). Deviations from the expectable environment in early childhood and emerging psychopathology. Neuropsychopharmacology (in press).
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K et al (2010). Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 167: 748–751.
Iyegbe C, Campbell D, Butler A, Ajnakina O, Sham P (2014). The emerging molecular architecture of schizophrenia, polygenic risk scores and the clinical implications for GxE research. Social Psychiatry Psychiatr Epidemiol 49: 169–182.
Jolin EM, Weller RA, Weller EB (2012). Occurrence of affective disorders compared to other psychiatric disorders in children and adolescents with 22q11.2 deletion syndrome. J Affect Disord 136: 222–228.
Jones W, Klin A (2013). Attention to eyes is present but in decline in 2-6-month-old infants later diagnosed with autism. Nature 504: 427–431.
Karg K, Burmeister M, Shedden K, Sen S (2011). The serotonin transporter promoter variant (5-HTTLPR), stress and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry 68: 444–454.
Kasari C, Kaiser A, Goods K, Nietfeld J, Mathy P, Landa R et al (2014). Communication interventions for minimally verbal children with autism: a sequential multiple assignment randomized trial. J Am Acad Child Adolesc Psychiatry 53: 635–646.
King BH, Hollander E, Sikich L, McCracken JT, Scahill L, Bregman JD et al (2009). Lack of efficacy of citalopram in children with autism spectrum disorders and high levels of repetitive behavior: citalopram ineffective in children with autism. Arch Gen Psychiatry 66: 583–590.
King MK, Jope RS (2013). Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav 12: 723–731.
Knapp M, Romeo R, Beecham J (2009). Economic cost of autism in the UK. Autism 13: 317–336.
Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E et al (1991). Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252: 1711–1714.
Kwan KY, Lam MM, Johnson MB, Dube U, Shim S, Rasin MR et al (2012). Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell 149: 899–911.
Lahat A, Lamm C, Chronis-Tuscano A, Pine DS, Henderson HA, Fox NA (2014). Early behavioral inhibition and increased error monitoring predict later social phobia symptoms in childhood. J Am Acad Child Adolesc Psychiatry 53: 447–455.
Liu HY, Potter MP, Woodworth KY, Yorks DM, Petty CR, Wozniak JR et al (2011). Pharmacologic treatments for pediatric bipolar disorder: a review and meta-analysis. J Am Acad Child Adolesc Psychiatry 50: 749–762 e739.
Loesch DZ, Bui QM, Dissanayake C, Clifford S, Gould E, Bulhak-Paterson D et al (2007). Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci Biobehav Rev 31: 315–326.
Lopez-Munoz F, Alamo C (2009). Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharmaceutical Design 15: 1563–1586.
Lord C, Risi S, Lambrecht L, Cook EH Jr., Leventhal BL, DiLavore PC et al (2000). The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 30: 205–223.
Maayan L, Correll CU (2011). Weight gain and metabolic risks associated with antipsychotic medications in children and adolescents. J Child Adolesc Psychopharmacol 21: 517–535.
Marshall M, Rathbone J (2011). Early intervention for psychosis. Cochrane Database Syst Rev CD004718.
Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR et al (2008). Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA 105: 13656–13661.
Messinger D, Young GS, Ozonoff S, Dobkins K, Carter A, Zwaigenbaum L et al (2013). Beyond autism: a baby siblings research consortium study of high-risk children at three years of age. J Am Acad Child Adolesc Psychiatry 52: 300–308.
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG et al (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74: 49–56.
Miklowitz DJ, O'Brien MP, Schlosser DA, Addington J, Candan KA, Marshall C et al (2014). Family-focused treatment for adolescents and young adults at high risk for psychosis: results of a randomized trial. J Am Acad Child Adolesc Psychiatry 53: 848–858.
Miklowitz DJ, Schneck CD, Singh MK, Taylor DO, George EL, Cosgrove VE et al (2013). Early intervention for symptomatic youth at risk for bipolar disorder: a randomized trial of family-focused therapy. J Am Acad Child Adolesc Psychiatry 52: 121–131.
Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA et al (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet 9: 1415–1423.
Miller G (2012). Alzheimer's research. Stopping Alzheimer's before it starts. Science 337: 790–792.
Molina BS, Hinshaw SP, Swanson JM, Arnold LE, Vitiello B, Jensen PS et al (2009). The MTA at 8 years: prospective follow-up of children treated for combined-type ADHD in a multisite study. J Am Acad Child Adolesc Psychiatry 48: 484–500.
Motiwala SS, Gupta S, Lilly MB, Ungar WJ, Coyte PC (2006). The cost-effectiveness of expanding intensive behavioural intervention to all autistic children in Ontario: in the past year, several court cases have been brought against provincial governments to increase funding for Intensive Behavioural Intervention (IBI). This economic evaluation examines the costs and consequences of expanding an IBI program. Healthcare Policy=Politiques de sante 1: 135–151.
Murphy DL, Lesch KP (2008). Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci 9: 85–96.
Nelson CA 3rd, Zeanah CH, Fox NA, Marshall PJ, Smyke AT, Guthrie D (2007). Cognitive recovery in socially deprived young children: the Bucharest Early Intervention Project. Science 318: 1937–1940.
Ozonoff S, Young GS, Belding A, Hill M, Hill A, Hutman T et al (2014). The broader autism phenotype in infancy: when does it emerge? J Am Acad Child Adolesc Psychiatry 53: 398–407.
Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L et al (2011). Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium Study. Pediatrics 128: e488–e495.
Pedersen MG, Mortensen PB, Norgaard-Pedersen B, Postolache TT (2012). Toxoplasma gondii infection and self-directed violence in mothers. Arch Gen Psychiatry 69: 1123–1130.
Pedersen MG, Stevens H, Pedersen CB, Norgaard-Pedersen B, Mortensen PB (2011). Toxoplasma infection and later development of schizophrenia in mothers. Am J Psychiatry 168: 814–821.
Pediatric OCD Treatment Study (POTS) Team (2004). Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA 292: 1969–1976.
Pine DS, Cohen P, Gurley D, Brook J, Ma Y (1998). The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry 55: 56–64.
Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S et al (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 45: 1150–1159.
Rogers SJ, Estes A, Lord C, Vismara L, Winter J, Fitzpatrick A et al (2012). Effects of a brief Early Start Denver model (ESDM)-based parent intervention on toddlers at risk for autism spectrum disorders: a randomized controlled trial. J Am Acad Child Adolesc Psychiatry 51: 1052–1065.
Ross EJ, Graham DL, Money KM, Stanwood GD (2015). Developmental consequences of fetal exposure to drugs: what we know and what we still must learn. Neuropsychopharmacology (in press).
Rutter M, Andersen-Wood L, Beckett C, Bredenkamp D, Castle J, Groothues C et al (1999). Quasi-autistic patterns following severe early global privation. English and Romanian Adoptees (ERA) Study Team. J Child Psychol Psychiatry 40: 537–549.
Sandler AD, Bodfish JW (2000). Placebo effects in autism: lessons from secretin. J Dev Behav Pediatr 21: 347–350.
Saxena R, Elbers CC, Guo Y, Peter I, Gaunt TR, Mega JL et al (2012). Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet 90: 410–425.
Scahill L, Aman MG, Lecavalier L, Halladay AK, Bishop SL, Bodfish JW et al (2013). Measuring repetitive behaviors as a treatment endpoint in youth with autism spectrum disorder. Autism (in press).
Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q et al (2012). A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci 32: 1803–1810.
Schmidt MJ, Mirnics K (2015). Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology (in press).
Shechner T, Rimon-Chakir A, Britton JC, Lotan D, Apter A, Bliese PD et al (2014). Attention bias modification treatment augmenting effects on cognitive behavioral therapy in children with anxiety: randomized controlled trial. J Am Acad Child Adolesc Psychiatry 53: 61–71.
Shen WW (1999). A history of antipsychotic drug development. Comprehensive psychiatry 40: 407–414.
Shonkoff JP, Garner AS, Committee on Psychosocial Aspects of C Family H Committee on Early Childhood A Dependent C et al (2012). The lifelong effects of early childhood adversity and toxic stress. Pediatrics 129: e232–e246.
Silverman JL, Yang M, Lord C, Crawley JN (2010). Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11: 490–502.
Siuta MA, Robertson SD, Kocalis H, Saunders C, Gresch PJ, Khatri V et al (2010). Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol 8: e1000393.
Strohl MP (2011). Bradley's Benzedrine studies on children with behavioral disorders. The Yale J Biol Med 84: 27–33.
Stronach S, Wetherby AM (2014). Examining restricted and repetitive behaviors in young children with autism spectrum disorder during two observational contexts. Autism 18: 127–136.
Suren P, Roth C, Bresnahan M, Haugen M, Hornig M, Hirtz D et al (2013). Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 309: 570–577.
Suri D, Teixeira C, Cagliostro MC, Mahadevia D, Ansorge MS (2015). Monoamine-sensitive developmental periods impacting adult emotional and cognitive behaviors. Neuropsychopharmacology (in press).
Taylor LE, Swerdfeger AL, Eslick GD (2014). Vaccines are not associated with autism: An evidence-based meta-analysis of case-control and cohort studies. Vaccine 32: 3623–3629.
Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD et al (2009). Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA 106: 2029–2034.
Tsapakis EM, Soldani F, Tondo L, Baldessarini RJ (2008). Efficacy of antidepressants in juvenile depression: meta-analysis. Br J Psychiatry 193: 10–17.
van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O'Collins V et al (2010). Can animal models of disease reliably inform human studies? PLoS Med 7: e1000245.
Walkup JT, Albano AM, Piacentini J, Birmaher B, Compton SN, Sherrill JT et al (2008). Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med 359: 2753–2766.
Warlaumont AS, Richards JA, Gilkerson J, Oller DK (2014). A social feedback loop for speech development and its reduction in autism. Psychological Sci 25: 1314–1324.
Warren Z, McPheeters ML, Sathe N, Foss-Feig JH, Glasser A, Veenstra-Vanderweele J (2011a). A systematic review of early intensive intervention for autism spectrum disorders. Pediatrics 127: e1303–e1311.
Warren Z, Veenstra-VanderWeele J, Stone W, Bruzek JL, Nahmias AS, Foss-Feig JH et al (2011b) Therapies for Children With Autism Spectrum Disorders. Agency for Healthcare Research and Quality (US): Rockville (MD). Report No. 11-EHC029-EF.
Wolff JJ, Gu H, Gerig G, Elison JT, Styner M, Gouttard S et al (2012). Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am J Psychiatry 169: 589–600.
Wright J (2014). Genetics: unravelling complexity. Nature 508: S6–S7.
Xie J, Bartels CM, Barton SW, Gu D (2013). Targeting hedgehog signaling in cancer: research and clinical developments. OncoTargets Therapy 6: 1425–1435.
Zhang J, Yang PL, Gray NS (2009). Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9: 28–39.
Acknowledgements
This work was supported, in part, by NIH grants MH094604 (JV-V) and HD073984 (JV-V and ZW).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Veenstra-VanderWeele, J., Warren, Z. Intervention in the Context of Development: Pathways Toward New Treatments. Neuropsychopharmacol 40, 225–237 (2015). https://doi.org/10.1038/npp.2014.232
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.232
This article is cited by
-
Assessing Social Communication and Collaboration in Autism Spectrum Disorder Using Intelligent Collaborative Virtual Environments
Journal of Autism and Developmental Disorders (2020)
-
VIA Family—a family-based early intervention versus treatment as usual for familial high-risk children: a study protocol for a randomized clinical trial
Trials (2019)
-
Developmental timing and critical windows for the treatment of psychiatric disorders
Nature Medicine (2016)
-
Antidepressive and BDNF effects of enriched environment treatment across ages in mice lacking BDNF expression through promoter IV
Translational Psychiatry (2016)
-
Brief Report: Evaluation of an Intelligent Learning Environment for Young Children with Autism Spectrum Disorder
Journal of Autism and Developmental Disorders (2016)
