
Abstract
Most drugs of abuse easily cross the placenta and can affect fetal brain development. In utero exposures to drugs thus can have long-lasting implications for brain structure and function. These effects on the developing nervous system, before homeostatic regulatory mechanisms are properly calibrated, often differ from their effects on mature systems. In this review, we describe current knowledge on how alcohol, nicotine, cocaine, amphetamine, Ecstasy, and opiates (among other drugs) produce alterations in neurodevelopmental trajectory. We focus both on animal models and available clinical and imaging data from cross-sectional and longitudinal human studies. Early studies of fetal exposures focused on classic teratological methods that are insufficient for revealing more subtle effects that are nevertheless very behaviorally relevant. Modern mechanistic approaches have informed us greatly as to how to potentially ameliorate the induced deficits in brain formation and function, but conclude that better delineation of sensitive periods, dose–response relationships, and long-term longitudinal studies assessing future risk of offspring to exhibit learning disabilities, mental health disorders, and limited neural adaptations are crucial to limit the societal impact of these exposures.
Similar content being viewed by others

INTRODUCTION
Substance use disorders among pregnant women continues to be a major public health concern, posing risk to the child’s development, and imposing socioeconomic burdens on society by increasing needs for medical and social services. Given the crucial roles for the major protein targets of drugs of abuse in shaping brain development (Bhide, 2009; Bonnin and Levitt, 2011; Frederick and Stanwood, 2009; Hohmann, 2003; Kater and Lipton, 1995; Lauder, 1993; Money and Stanwood, 2013; Stanwood and Levitt, 2004), it should not be surprising that fetal drug exposures have been linked to a wide variety of brain deficits. In this review, we will focus on: (1) what is currently known about the likely pattern of substance use among pregnant or women of childbearing age; (2) the cellular and molecular pathways by which prenatal drug exposure may influence structural and functional brain development; (3) current studies on outcomes of exposed individuals across various areas of functioning (neurobiology, physical growth, intelligence, executive functioning, behavior, and psychopathology); (4) reviews of current experimental animal models, and (5) current limitations in understanding and potential avenues for future research. While beyond the scope of the current review, untreated drug abuse/addiction also typically coincides with poor nutrition and prenatal care, which increases the risk of obstetric complications and disrupted developmental processes in the fetus. Thus, beyond the specific developmental biology of individual drugs, there are additional common factors that can produce deficits in neurodevelopmental trajectories.
Recent data suggest that nearly 25 million Americans aged 12 or older are current illicit drug users; this estimate represents 9.2 percent of the population. Illicit drugs include marijuana/hashish, cocaine (including crack), heroin, hallucinogens, inhalants, or prescription-type psychotherapeutics used non-medically (Substance Abuse and Mental Health Services Administration, 2013). Current illicit drug use among pregnant women aged 15–44, has remained constant at 5.9% despite efforts of prevention and education programs (Substance Abuse and Mental Health Services Administration, 2013). In fact, the pattern of rates of current illicit drug use among young adolescent females has grown to where they are now more likely than males in their age group to be current non-medical users of psychoactive drugs.
Legal drugs such as alcohol and nicotine also represent a significant hurdle regarding unintended effects on the fetus. The 2012 NSDUH also found that among pregnant women aged 15–44, the prevalence of reported current alcohol and cigarette usage rates has not changed substantially in the past decade. Although the US population as a whole is smoking less, the past month cigarette use among pregnant women aged 15–44 has held at ∼16–17% over the past 2 years (as compared with 18% in 2002–2003). An annual average of ∼8.5% also reports current alcohol use during pregnancy. As described below, legality of a drug does not necessarily correlate with its safety profile, and as researchers and clinicians, we need better therapies and educational strategies for pregnant and/or breastfeeding women.
Different drugs and biological processes are often modeled in different animal species, each with its own unique developmental timeline (Figure 1) and advantages/disadvantages. This greatly complicates translatability to humans. Rodents (mice, rats) and lagomorphs (rabbits) are born relatively early, as compared with humans, and thus those offspring must be treated with drugs postnatally if the aim is to model third trimester human exposure (Figure 1). However, this omits the transplacental transfer that is inherent in human fetal drug exposure. Issues of accurate dose levels, route of administration, and pharmacokinetics/bioavailability are also often underappreciated in animal models.

Major neurodevelopmental events across species. Schematic diagram that aligns human brain development with several animal models (monkey, rabbit, rat, and mouse) often used in studies of fetal drug exposure. Note in particular that the rodent equivalent of third trimester fetal development occurs postnatally.
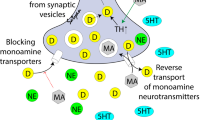
Moreover, drugs can alter fetal development through a wide variety of mechanisms (Figure 2). For example, if the drug crosses the placenta (and the vast majority of drugs of abuse do cross), then it can directly act on its molecular target in the fetus. Drugs can also act directly on the uterus and/or placenta. These effects would include altering placental secretory activity or uteroplacental blood flow, for example. Finally, the drug can produce effects on the mother's physiology that may secondarily influence the fetus, such as increased secretion of stress hormones or altered maternal health behaviors attributable to the mother’s addiction. And although it is beyond our scope to review it here, it has recently become clear that paternal exposures, to drugs such as cocaine, during spermatogenesis, can also influence offspring brain development and neurobehavioral development through epigenetic mechanisms, at least in animal models (Killinger et al, 2012; Rodgers et al, 2013; Vassoler et al, 2013). This topic is thus extremely complicated; nevertheless, we will do our best to review both animal models and clinical and imaging data from longitudinal human cohorts following fetal exposure to specific drugs of abuse.

Biological targets of fetal drug exposures. Drugs of abuse not only target the developing fetal brain directly, but can exert effects through a variety of organs with the mother, including the uterus, placenta, heart, lungs, and brain.
PSYCHOSTIMULANTS
Methamphetamine
Methamphetamine (METH) is metabolized in rat and human livers to the main metabolites, amphetamine, and 4-hydroxymethamphetamine (Caldwell et al, 1972) (prenatal amphetamine exposure discussed below). Due to the addition of a methyl group, METH has a higher lipophilicity than amphetamine, allowing more rapid transport of the drug across the blood–brain barrier (Barr et al, 2006). METH has a high potential for abuse and addiction compared with other amphetamine-type stimulants, affecting the serotonin (5-HT) plasma membrane transporter (SERT) and activating the psychological reward system by triggering a cascade of the massive release of dopamine (DA) in the brain. This release of DA occurs by multiple mechanisms including: binding to reuptake transporter (DAT), the displacement of vesicles and inhibition of monoamine oxidase, and enhancing DAT-mediated reverse transport of DA transport across the plasma membrane (Scott et al, 2007). These targets of METH appear by mid-gestation in the fetal brain (Bhide, 2009; Frederick and Stanwood, 2009; Malanga and Kosofsky, 1999; Money and Stanwood, 2013).
The use of METH in the United States and in other parts of the world still remains a serious public health concern. According to the United Nations, 1.3% of the general population in Central and North America use amphetamine-type stimulants (UNODC, 2013). There are few studies that have surveyed the prevalence of METH use among pregnant women. The only large-scale investigation to report METH prevalence for pregnant women has been the Infant Development, Environment, and Lifestyle (IDEAL) study. They found that ∼5% of women self-reported METH use during their pregnancy (Arria et al, 2006) in areas of the United States where METH use was of high concern. Furthermore, over 40% of the women enrolled in the IDEAL study continued using the drug during their third trimester, and approximately half did not significantly change their personal use during the course of the pregnancy (Della Grotta et al, 2010).
Despite the fact that METH has been utilized by pregnant women for many years, relatively little is known about the effects of METH during early infancy and even less is known about the long-term effects. The most common effects noted in newborns indicate that METH use is associated with growth restriction, decreased weight, length, and head circumference (Little et al, 1988; Nguyen et al, 2010; Smith et al, 2003; Smith et al, 2006b). Echoencephalography has revealed congenital anomalies in exposed neonates include cardiac anomalies, cranial abnormalities, and abnormal brain development closely resembling those in ill, asphyxiated infants (Dixon and Bejar, 1989). Mild withdrawal symptoms have also been noted in newborns (Oro and Dixon, 1987; Smith et al, 2003), although these symptoms are not as common. In animal models, increased DNA oxidation and postnatal functional deficits in motor coordination were documented in neonatal METH-exposed mice (Jeng et al, 2005).
Several recent studies have examined the effects of METH exposure on childhood growth patterns and behavior after birth. Focusing on physical growth patterns, Zabaneh et al (2012) found children exposed prenatally to METH have a modest decrease in height during the first 3 years of life with no observed difference in weight, head circumference, or weight-for-length trajectories. Prenatal METH exposure also is associated with significantly reduced caudate nucleus volume and cortical thickness increases in perisylvian and orbital–frontal cortices (Derauf et al, 2012; Zabaneh et al, 2012). Diffusion tensor imaging (DTI) suggests lower diffusion and higher fractional anisotropy in METH-exposed children at 3–4 years of age, indicating that fetal METH may alter white matter tracts (Cloak et al, 2009). In a magnetic resonance spectroscopy (MRS) study, METH-exposed 3–4 year-old children exhibited higher total creatine, N-acetyl compounds, and glutamate+glutamine concentrations in the frontal white matter, but lower myoinositol and myoinositol/creatine in the thalamus (Chang et al, 2009). The higher metabolite concentrations of N-acetyl compounds, total creatine, and glutamate+glutamine suggest a higher neuronal density or cellular compactness in the white matter, whereas lower myoinositol suggests lower glial content in the thalamus. Furthermore, the METH-exposed children performed significantly worse on a visual motor integration task, which correlated with lower myoinositol in the thalamus (Chang et al, 2009). In another magnetic resonance imaging (MRI) study on children ranging from 3 to 16 years of age, exposed children scored lower on measures of visual motor integration, attention, verbal memory, and long-term spatial memory. There were no differences among the groups in motor skills, short delay spatial memory, or measures of nonverbal intelligence. Despite comparable whole brain volumes in each group, the METH-exposed children appeared to have subtle but significant decreases in size or volume in certain brain regions, including the putamen, globus pallidus, and hippocampus. These reductions correlated with poorer sustained attention and delayed verbal memory (Chang et al, 2004). It is important to note that although the study had an impressive age range, sample sizes were limited.
Concentrating on long-term motor skills after birth, Smith et al (2011) observed a subtle METH exposure effect on fine motor performance at 1 year, with the poorest performance observed in the most heavily exposed children. However, by 3 years of age, no differences in fine motor performance were observed. These findings suggest METH exposure has modest motor effects in the first year of life, but that this may be mostly resolved by 3 years of age. The IDEAL study found that prenatal METH exposure was associated with child externalizing behavioral problems at 5 years and parenting stress and psychological symptoms experienced by primary caregivers were associated with increased child behavioral problems; indicating these effected children may have more difficulties negotiating the increasing complex academic and social demands of school-age children (LaGasse et al, 2012; Twomey et al, 2013).
Animal models may give us insight and provide correlation to these studies examining the effects of prenatal METH exposure and if these effects will remain into adulthood. Results from mouse exposure to METH during a time period equivalent to the third trimester of human fetal gestation (see Figure 1) impaired weight gain, reduced novel location recognition, and impaired novel object recognition in both male and female mice during adolescence. In rats, neonatal exposure to METH produces deficits in latency and memory in a Morris water maze (Vorhees et al, 2000). Both prenatal and neonatal exposure also caused impaired spatial learning (Slamberova et al, 2005; Williams et al, 2003b; Williams et al, 2003d; Williams et al, 2002) and neonatal exposure altered the adrenal response to a forced swim stressor, suggesting that the adrenal output during learning may contribute to the spatial learning deficits (Williams et al, 2003a). Rats exposed to METH in utero showed changes in the mesolimbic dopaminergic system and were more sensitive to the administration of the acute dose of METH in adulthood (Bubenikova-Valesova et al, 2009). This indicates that offspring exposed to METH in utero could be more sensitive to METH and potentially to other psychostimulants (Bubenikova-Valesova et al, 2009).
Although relatively little is known about the effects of METH during early infancy or the following long-term effects, recently there have been studies attempting to examine children exposed to METH in utero. Further studies are important to aid in prevention programs and treatment for these individuals. Comparing the effects of prenatal METH exposure on infant and childhood growth between the United States and New Zealand demonstrated that the effects of prenatal METH differed across countries (Abar et al, 2013). In the study, the prenatal METH-exposed children in New Zealand fared better than exposed children in the United States. In addition, according to the United Nations, 1.3% of the general population in Central and North America use amphetamine-type stimulants (UNODC, 2013). This reported use is at higher levels than the global average of 0.7% of the general population (UNODC, 2013). These studies suggest that the United States needs improved prevention programs, better pre- and postnatal treatments, and caregiver support for this exposed population.
Amphetamine
Amphetamines (alpha-methyl-phenethylamine) (AMPH), commonly known as ‘Speed’, continue to be widely used by women of childbearing age. The users are either legally prescribed AMPHs for medical reasons or are nonmedical users. For example, attention deficit hyperactivity disorder (ADHD) affects ∼5% of adults (Fayyad et al, 2007) in the United States. Although ADHD is less common in females than males, a significant percentage of women of childbearing age are thus likely prescribed AMPH or other stimulants as a treatment for ADHD. The prevalence of women of childbearing age using non-medical AMPH is currently unknown, and even less is known about the frequency of use during pregnancy. Further studies are required to determine the true frequency of illegal AMPH-exposed pregnant and lactating women, so that appropriate care can be provided for mother and her family.
After prenatal exposure, AMPH has been detected in human umbilical cord (Jones et al, 2009), plasma (Dearlove and Betteridge, 1992), and placenta as early as the first trimester (Joya et al, 2010). The cellular actions of AMPH are nearly identical to METH (see section above), including increasing the levels of norepinephrine (NE), DA, and 5-HT in the synaptic cleft via transporter reuptake inhibition. This increased availability allows the monoamines to act upon post-synaptic receptors. The use of AMPHs during pregnancy increases the risk of adverse effects on the outcome of pregnancy, such as placental hemorrhage (Figure 2). This is mediated, at least in part, by stimulation of contractions in the uterus and by targeting NET and SERT in the human placenta (Cordeaux et al, 2008; Ramamoorthy et al, 1995). These actions have been hypothesized to contribute to preterm labor associated with AMPH exposure (Cordeaux et al, 2008). To date, fetal AMPH exposure has not been proven to be directly teratogenic; however, in primary human cell cultures, AMPH reduced folic acid uptake and this could potentially lead to placental and/or fetal toxicity (Keating et al, 2009). Animal studies revealed at a prepubertal age, an enhancement of D1 receptors in the dorsal striatum and nucleus accumbens (NAc) and a decrement of the D3 receptors in NAc, olfactory tubercle, and the islands of Calleja. In contrast, at a postpubertal age, the authors instead measured an increase in the levels of DAT in the NAc and striatum, and a decrease in D2 receptor expression in the NAc shell. In addition, acute AMPH induces a marked decrease in locomotor activity in rats following prenatal AMPH exposure (Flores et al, 2011). These developmental and behavioral changes in animal models associated with in utero AMPH exposure provide insights to the mechanisms causing changes in affective, behavioral, and cognitive outcomes in exposed children.
A meta-analysis of 10 studies of low-to-moderate risk of bias submits that AMPH exposure in pregnancy is associated with higher odds of preterm birth, low birth weight, and small size for gestational age (Ladhani et al, 2011). Birth weight as a continuous variable was also found to be significantly lower among exposed women. Gestational age, however, was not found to be significantly different (Ladhani et al, 2011). The most extensive follow-up data on affective, behavioral, and cognitive outcomes following prenatal AMPHs are provided by Swedish researchers who tracked a cohort of 65 AMPH-exposed children from birth to age 14. They reported in children with continuous AMPH exposure throughout gestation, a variety of adverse physical, cognitive, emotional, and social effects, including increased prevalence of ADHD, aggression, and learning difficulties attributed to deficits in attention, memory, and motivation (Eriksson et al, 1994; Eriksson et al, 2000). Furthermore, a relationship between head circumference at birth and at 1 year of age predicted language and mathematics proficiency at 14 years of age (Eriksson et al, 2000). As with its parent drug METH, further studies on prenatal AMPH exposure is needed to better understand effects from prescribed AMPHs or non-medical users (Oei et al, 2012).
3,4-Methylenedioxymethamphetamine
3,4-Methylenedioxymethamphetamine (MDMA), commonly known as Ecstasy, is a derivative of AMPH that has both stimulant and hallucinogenic properties. It acts as a powerful releasing agent of 5-HT, NE, and DA and also acts as a reuptake inhibitor of their high-affinity transporters (de la Torre et al, 2004; Rudnick and Wall, 1992). MDMA is actively transported through the plasma membrane and can then inhibit the vesicular monoamine transporter, resulting in increased concentrations of 5-HT, NE, and DA in the cytoplasm. MDMA can also directly bind a number of receptors with moderate affinity, including α2-adrenergic (adrenaline) and 5-HT2A receptors (de la Torre et al, 2004). A single MDMA injection to pregnant rat dams results in MDMA transfer into the fetal compartment (Campbell et al, 2006). However, emerging data also suggests key roles for maternal and placental 5-HT in regulating brain development (Bonnin et al, 2011; Bonnin and Levitt, 2011) (Figure 2).
In the United States, data from the 2012 National Survey on Drug Use and Health estimate that about 6.2 percent of individuals aged 12 or older had used Ecstasy at least once in their lifetime. In addition, ∼900 000 individuals tried Ecstasy for the first time in 2012. Although data from pregnant women was not presented, the prevalence of at least once in lifetime Ecstasy use in 2012 saw a significant increase from past-year use, and past-year use for eighth through twelfth graders remained stable (UNODC, 2013).
To date, there are only a few studies examining prenatal exposure to MDMA in humans, despite concerns about its potential harmful effects to the fetus (Ho et al, 2001; McElhatton et al, 1999; Singer et al, 2012; van Tonningen-van Driel et al, 1999). Very little is known about the characteristics of pregnant women who use Ecstasy. Using data collected regarding women contacting the Motherrisk Helpline in Toronto (Ho et al, 2001), MDMA users tended to be younger, single, Caucasian, binge drinkers, and had a higher prevalence of psychiatric symptoms. Most of these users discontinued MDMA once pregnancy was known, but because the recruitment was based on contact initiated by the mothers, the sample likely overrepresented more ‘motivated’ women. The few available studies demonstrate that pregnant MDMA users exhibit a clustering of reproductive risk factors that contribute to neurobehavioral and teratological outcomes. A retrospective report of 136 babies exposed to Ecstasy in utero noted premature births, a significantly increased risk of congenital defects, cardiovascular anomalies, and musculoskeletal anomalies (McElhatton et al, 1999). Another study in the Netherlands reported congenital cardiac malformation and spontaneous abortions (van Tonningen-van Driel et al, 1999). A recent neurobehavioral outcome study suggests that prenatal MDMA exposure predicts poorer infant mental and motor development at 4 and 12 months of age in a dose-dependent manner (Singer et al, 2012).
Only a handful of animal models of pre- and perinatal MDMA exposure have been developed as well. A rat study of prenatal MDMA exposure (E14–E20) described increases in dopaminergic fibers in the prefrontal cortex, striatum, and NAc—areas critical in novelty responses, reward, attention, and motor behavior (Thompson et al, 2009b). It also appears to increase NE fiber density in the prelimbic region of the prefrontal cortex and in the hippocampus (Thompson et al, 2012).
Further studies suggest that developmental MDMA, particularly during a time analogous to the early third trimester, can produce persistent reductions in DA and 5-HT metabolites (Koprich et al, 2003a; Koprich et al, 2003b). Meyer et al (2004) determined that MDMA exposure from P1 to P4 can decrease 5-HT levels in the hippocampus on P25 and P60, and suggested that MDMA treatment stimulates apoptotic cell death during early postnatal development. Significant reductions in 5-HT fiber density were observed in the cerebral cortex with a hyperinnervation in the caudate–putamen and NAc in 9–month-old animals, demonstrating enduring effects (Meyer et al, 2004). The early hippocampal changes were not observed in adulthood, suggesting a recovery of damaged serotonergic fibers following neonatal exposure. The Meyer lab further demonstrated that developmental MDMA administration can lead to long-lasting functional abnormalities including enhanced sensitivity to the drug later in life (Piper et al, 2009; Piper and Meyer, 2006).
Other studies have shown few effects of MDMA exposure in developing animal models. This could possibly be because of time period of treatment—embryonic versus postnatal—or doses (once again, please see Figure 1 for a description of human and animal development timelines). For example, when rats were exposed to MDMA twice a day from E12 to E15, no differences were seen in hippocampal levels of 5-HT, DA, NE, or metabolites when assayed on P21 (Winslow and Insel, 1990). In addition, no changes in 5-HT or DA levels in various brain regions were observed in rat offspring whose mothers were exposed to 20 mg/kg MDMA once per day every other day beginning at E6 until birth (Aguirre et al, 1998).
As adults, prenatal MDMA-exposed rats exhibit increased activity, risk-taking behavior, and altered spatial learning; however, it did not affect their feeding or food reward, or alter cocaine self-administration behaviors or locomotor responses (Thompson et al, 2009b). Neonatal administration of MDMA also produces impaired path integration learning (Broening et al, 2001; Vorhees et al, 2004; Williams et al, 2003c). Neonatal MDMA exposure alters the release of DA and 5-HT at P70 in the striatum and hippocampus, and reduces sucrose preference (an index of reward systems) at P70 (Galineau et al, 2005).
The detailed mechanisms through which prenatal/perinatal MDMA exposure produces alterations in neurodevelopmental trajectory are thus not fully understood. Heavier use has been associated with impaired motor and intellectual development in infants, but whether there are long-lasting changes in neurobehavioral outcomes is not yet known. A major concern is possible serotonergic dysfunction produced by repeated and/or high doses of the drug. A recent animal report suggests that that the simultaneous abuse of alcohol and Ecstasy during pregnancy, even for short periods of time, may cause much more significant abnormalities in neurocognitive development than either drug alone (Canales and Ferrer-Donato, 2014). Given the widespread recreational use of MDMA (Ecstasy), pregnant women should be cautioned about possible developmental effects in offspring. Animal models need to be designed to include relevant doses and human developmental periods to best ascertain the developmental effects of MDMA (for an excellent review, see Skelton et al (2008)).
Cocaine
Currently the percentage of women of childbearing age consuming cocaine is unknown and less information is known about the current frequency of use during pregnancy. According to the latest figures from (UNODC, 2013), cocaine was the third largest abused illicit drug (Substance Abuse and Mental Health Services Administration, 2013) within the past year. Among women ages 12 and older, including women of childbearing age, 0.3% were current users of cocaine (Substance Abuse and Mental Health Services Administration, 2013). Thus, cocaine use in women continues to be a major public health concern. It has been noted that prenatal cocaine exposure alone accounts for over 26 million dollars per year in special education services in the United States (Levine et al, 2008).
Studies of cocaine in animal models, using a variety of species, have demonstrated cocaine easily crosses both the placenta and blood–brain barrier and can have teratogenic effects on the developing fetus (Mayes, 1994). The negative outcomes and effects from neonatal exposure to cocaine can result from the pathophysiology originating from three possible pathways; first, cocaine directly inhibits the reuptake of multiple monoamines at the presynaptic junction, leading to increased concentrations of DA, 5-HT, and NE within the synaptic cleft. Also, neonatal cocaine exposure has been attributed to indirect vasoconstrictive effects and higher levels of activation in the catecholaminergic system (Nassogne et al, 1998). In addition to the neurochemical and vascoconstrictive effects of cocaine on fetal development, cocaine may also act by altering genetic programming (Lester and Padbury, 2009).
Clinical reports of the impact of prenatal cocaine have been varied; some suggest global and severe physical malformations, others document specific deficits in physical, cognitive, and emotional development, and yet other studies indicate no effects. It is important to note that the magnitude of these effects depend upon the dosage, gestational timing, duration of exposure, and/or postnatal care. Some of the disparities in the human literature arose from the problem that many of the early studies were poorly controlled and produced media hysteria around so-called ‘crack babies’. Nevertheless, well-controlled studies have demonstrated that prenatal cocaine exposure does in fact affect fetal physical growth, and results in an increase of premature birth, and generalized growth retardation—including decreased birth weight, shorter body length, and smaller head circumference (Bigsby et al, 2011; Covington et al, 2002; Gouin et al, 2011; Mayes et al, 2003). Abnormal infant behavioral outcomes have also been documented; these outcomes include abnormalities related to lower arousal, poorer quality of movement and self-regulation, higher excitability, jitteriness, and more non-optimal reflexes (Lester et al, 2002; Richardson et al, 2008; Singer et al, 2000; Tronick et al, 2005). Studies have revealed that the behavioral outcomes observed at birth continue and sometimes worsen after 12 months of age (Bigsby et al, 2011; Chiriboga et al, 2007; Mayes et al, 2003; Richardson et al, 2008).
Growth restriction among prenatal cocaine exposure children has been documented to continue well past infancy (Minnes et al, 2006) and may persist in children as old as 10 years of age (Covington et al, 2002; Richardson et al, 2013). While consistency exists in the reports of reduced fetal growth and development, some follow-up studies at later ages have suggested catch-up growth. Shankaran et al (2011) found that children exposed to prenatal cocaine were similar in weight to non-exposed children at 6 years of age. Interestingly, children who had been exposed to high levels of prenatal cocaine, and were born full-term, were reported to have a higher body mass index and blood pressure changes. These variable outcomes of prenatal cocaine exposure on physical growth and development are in part the result of important covariates such as amount of drug use and timing throughout pregnancy. Other possible variables are disturbances in physiological regulation, such as the respiratory sinus arrhythmia and cardiac development (Finger et al, 2014). Studies in animal models also provide evidence for a programming effect resulting in detrimental long-term changes to the heart induced by fetal cocaine exposure (Meyer and Zhang, 2009). Overall, additional long-term follow-up studies of children with neonatal cocaine exposure and its effects on health outcomes are needed to help identify mechanisms that result in abnormal growth and development.
Although prenatal cocaine exposure alone does not appear to lower global intelligence, there is consistent evidence of poor cognitive performance in language skills, behavior, and executive functioning. Longitudinal studies have reported that prenatal cocaine is associated with impaired language development through early adolescence (Bandstra et al, 2011; Lewis et al, 2007; Lewis et al, 2011). However, some studies suggest a general improvement in receptive language starting in adolescence through age 17 (Betancourt et al, 2011). Furthermore, adoption or foster care appears to enrich the children’s linguistic environment and helps to protect children against some language delays (Lewis et al, 2011).
Long-term studies have described varying effects on executive function in exposed children. Executive function is a set of mental processes for the management of cognitive operations that include attention, behavior, cognition, working memory, and information/problem solving. Richardson et al (2013) found that first trimester cocaine exposure has been associated with less sociability, more withdrawn behavioral problems, more anxious/depressed behaviors and symptoms in the children. These behaviors may be precursors of later psychiatric problems. By caregiver reports, higher cocaine use was associated with disruptive behaviors including aggression and delinquent behavior at 9 years of age (McLaughlin et al, 2011). Moreover, caregiver reports indicate overall issues of executive function in 12 year-old children with higher amounts of prenatal exposure; in particular, females had greater problems with initiation activities, working memory, and organization (Minnes et al, 2014). Carmody et al (2011) further documented cocaine exposure affected the attention and inhibitory control performance of males, but not females, in children at ages 6, 9, and 11. The neurobiological mechanisms leading to gender-dependent differences in disrupted executive function following prenatal cocaine are not currently understood; however, this is an important consideration for future investigation.
These longitudinal studies have described varying effects on executive function in exposed children; however, it is also important to know the structure, function, and pharmacology of the brain that is possibly being altered by the drug exposure. Neuroimaging—for example, structural MRI, functional magnetic resonance imaging (fMRI) and DTI—is valuable in examining the underlying neural circuitry and connectivity. Recent reports describe long-term structural alterations in brain regions such as cortical and limbic regions (Rando et al, 2013). Lower fractional anisotropy in the right arcuate axons and higher mean diffusivity in the splenium of the corpus callosum has also been observed (Lebel et al, 2013). Cortical thickness of the right dorsolateral prefrontal cortex appears to be thinner in adolescents following prenatal cocaine (Liu et al, 2013), and cocaine-exposed adolescents have reduced ventral prefrontal cortex activation in response to increased memory load. Reduced structural connectivity between the ventral prefrontal cortex and the amygdala was also observed through DTI measurements (Li et al, 2013).
Overall, fetal cocaine exposure can affect fetal and long-term growth patterns, as well as cause language deficits, behavior defects, and executive functioning abnormalities. Animal models, across multiple species, give us further insight on the pathophysiological mechanisms that are possibly occurring during the developmental changes and are not accessible to study in vivo. Animal models confirm that prenatal cocaine exposure results in specific and long-lasting behavioral, cellular, and molecular changes (Bhide, 2009; Dow-Edwards, 2011; Harvey, 2004; Lidow et al, 2003; Mactutus et al, 1994; Malanga and Kosofsky, 1999; Mayes, 2002; Stanwood and Levitt, 2004). Moreover, animal models of prenatal exposure have disrupted cortical neurogenesis and migration during and after birth, reduction of neuronal numbers and density in the cortex, and differences in dopaminergic function (Crandall et al, 2004; Hamilton et al, 2010; McCarthy and Bhide, 2012; McCarthy et al, 2012; Ren et al, 2004). Specific anatomical defects include aberrant growth of dendrites of cortical projection and interneurons, suggesting disruption of local circuitry, and behavioral abnormalities that involve learning and stereotypic motor behavior (Mayes, 2002; Stanwood et al, 2001b; Thompson et al, 2005). Data suggest that the equivalent of the second trimester may be the most ‘sensitive period’ for the actions of fetal cocaine (Stanwood et al, 2001a). Alterations in the fetal development of the monoaminergic system can affect short- and long-term attention and cognitive development. Perhaps a point of origin for the disturbances from the effects of cocaine is the reduction of the coupling of the D1 receptor to its G protein-coupled receptor (Jones et al, 2000; Stanwood et al, 2005; Zhen et al, 2001). This uncoupling is sustained into adulthood and is specific for D1 receptors. Behavioral changes in response to cocaine exposure have also been reported in multiple animal models, including deficits in attention and emotional reactivity (Gabriel et al, 2003; Garavan et al, 2000; Harvey et al, 2001; Morrow et al, 2002a, 2002b; Stanwood and Levitt, 2003; Thompson et al, 2005; 2009a, ). These findings correspond with the human clinical literature, which reports disturbances in both attention and emotion regulation in children exposed prenatally to cocaine. Gestational cocaine also increases the sensitivity to the conditioned rewarding effects of cocaine in male rats and modestly affected females (Dow-Edwards et al, 2014), indicating alterations in the development of reward circuits.
This brief discussion of prenatal exposure to cocaine clearly emphasizes the complexity of determining the pathophysiological mechanisms and associated risk factors. Given the consistently large number of affected individuals, there is a constant need for furthering research and medical, mental health, and educational services for this impacted population.
OPIOIDS
Opiate receptors are G protein-coupled receptors and fall into three groups: mu, delta, and kappa. In situ hybridization of the adult rat brain identified mRNA for all three receptors throughout the central nervous system as well as numerous peripheral tissues (Wittert et al, 1996). Opioid receptor expression (Barg and Simantov, 1989; Lenoir et al, 1984) and endogenous opioid concentrations in the fetus and neonate differ from that in adults (Barg and Simantov, 1989). Thus, administration of opiates in utero may have more distinctive effects compared with adult exposure (Pertschuk et al, 1977).
Illicit opiate use has been steadily increasing in the past decade and a large part of this increase is in the 18–25 age bracket, which includes women of reproductive age (Substance Abuse and Mental Health Services Administration, 2013). The probability of preeclampsia, premature labor and rupture of membranes, placental insufficiency, abruptio placentae, intrauterine growth retardation, and intrauterine death increases greatly with illicit opiate use during pregnancy (Bashore et al, 1981; Hulse et al, 1998; Kaltenbach et al, 1998). Even with a successful labor and delivery, neonates often have low birthweight and smaller head circumference as well as experience symptoms of opiate withdrawal (Binder and Vavrinkova, 2008; Hunt et al, 2008; Kandall et al, 1976). Some clinical studies have also suggested an increased prevalence of heart defects, autonomic dysregulation (Paul et al, 2014), nystagmus (Gupta et al, 2012), and strabismus (Gill et al, 2003) in children exposed prenatally to opiates. At the pre- and elementary school ages, these children show motor and cognitive impairments (Bunikowski et al, 1998; Guo et al, 1994; Hunt et al, 2008), inattention (Hickey et al, 1995; Ornoy et al, 1996), hyperactivity (Ornoy et al, 1996), and an increase in ADHD when exposed prenatally to heroin (Ornoy et al, 2001). The damage of prenatal opiate exposure is debilitating and long lasting, and physicians must continue to track cohorts of exposed children to further understand the impact into adulthood.
Similar to human clinical studies, rodents exposed to heroin or morphine have a lower birthweight (Eriksson and Ronnback, 1989; Lu et al, 2012; Zagon and McLaughlin, 1977b, 1977c) and impaired learning and memory (Steingart et al, 2000a; Wang and Han, 2009). Numerous structural and functional alterations have been found that could underlie the effects of prenatal opiates on cognition, including perturbations in dendritic length, synaptic plasticity, neuronal proliferation, and cholinergic function. Prenatal heroin or morphine exposure decreases dendritic branch length in layer II/III pyramidal neurons in somatosensory cortex, which is thought to be specific to opiate receptor activation, as it can be blocked by co-administration of an opiate receptor antagonist (Lu et al, 2012; Ricalde and Hammer, 1990). Long-term potentiation, long-term depression, and proteins associated with synaptic transmission are all attenuated with perinatal morphine exposure (Villarreal et al, 2008; Yang et al, 2006). Decreased proliferation in the developing striatum (Harlan and Song, 1994) and increased apoptosis in dopaminergic cell cultures and the hippocampus have been observed with perinatal heroin or morphine exposure (Oliveira et al, 2003; Oliveira et al, 2002; Svensson et al, 2008; Wang and Han, 2009).
The Yanai lab has provided substantial evidence to suggest that the deficits in spatial learning and memory may be tied to hippocampal cholinergic alterations. They found increased hippocampal levels of a cholinergic receptor (muscarinic M1 receptor) and the choline transporter with perinatal heroin (Steingart et al, 2000a; Steingart et al, 2000b). In addition, they observed altered levels and activity of protein kinase C—a signaling protein downstream of the M1 receptor- and enhanced inositol phosphate induction by cholinergic agonists (Steingart et al, 2000a; Steingart et al, 2000b; Yaniv et al, 2004). Whether a cholinergic deficit is found in cohorts of opiate-exposed children is unknown.
Perinatal heroin and morphine exposure also disrupt maturation of the opiate receptor system. Postnatal morphine exposure decreases mu opioid receptor binding in the striatum, NAc, amygdala, hypothalamus, and spinal cord (Hammer et al, 1991; Kirby, 1983; Tempel, 1991). Perinatal morphine exposure also induces morphine tolerance (Chiang et al, 2010; Eriksson and Ronnback, 1989; Hovious and Peters, 1984), although an increased sensitivity to morphine analgesia has been reported in female offspring (Arjune and Bodnar, 1989). Aroyewun and Barr (1982) proposed that postnatal morphine also accelerates the maturation of some aspects of opiate-dependent behaviors, such as opiate antagonist-induced anorexia, that normally only occurs after P14. Postnatal morphine exposure accelerated the appearance of this behavior to P10 and 12 (Aroyewun and Barr, 1983), but the observed hypophagia could also have been induced by the precipitation of opiate withdrawal. In addition, perinatal morphine or heroin has also been shown to alter sexual behavior (Vathy and Katay, 1992), NE turnover and release (De Vries et al, 1991), neuroendocrine function (Litto et al, 1983), and several other important structures and processes that this review cannot cover (for a comprehensive review of the developmental effects of illicit opiates see Slamberova (2012)).
Opiate Maintenance Therapies
The American Academy of Pediatrics and American College of Obstetricians and Gynecologists recommend opioid maintenance therapy as the first line of treatment for opioid dependence during pregnancy (ACOG Committee, 2012). Untreated illicit opiate use is associated with poor prenatal care, nutrition, and fetal health, which is improved with opioid maintenance therapy (Binder and Vavrinkova, 2008; Kandall et al, 1976; Maas et al, 1990). Initiation of an effective opioid maintenance therapy facilitates better prenatal care, decreased illicit use of opiates and other drugs, and a continuous dosing regimen that prevents maternal/fetal withdrawal. Opiate withdrawal is not recommended during pregnancy unless the mother refuses opiate maintenance therapy due to increased likelihood of relapse (ACOG Committee, 2012). Animal studies have demonstrated in utero withdrawal to increase fetal activity and perinatal mortality (Kirby and Holtzmann, 1982; Kuwahara and Sparber, 1981; Lichtblau and Sparber, 1981).
Opioid maintenance therapies are not without substantial risk, as they can cross the placenta and alter development. Withdrawal from illicit or prescribed opiates incites Neonatal Abstinence Syndrome in neonates, which often requires treatment with morphine, diluted tincture of opium, or methadone with or without diazepam (Finnegan et al, 1975; Jansson et al,2009). However, incidence, peak severity score, duration, and length of required hospital stay due to Neonatal Abstinence Syndrome symptoms are less severe in neonates born to women following medically controlled maintenance therapies compared near-term mothers still using illicitly (Binder and Vavrinkova, 2008; Kandall et al, 1976; Maas et al, 1990).
Methadone
Methadone is considered the current gold standard opioid maintenance therapy for pregnant women. It is a long-acting full mu opioid receptor agonist that is distributed as a daily dose only at licensed methadone clinics; allowing a tightly regulated, appropriately titered dosing regimen, and decreased abuse. Even though methadone reduces symptoms compared with illicit opiate use, clinical cohorts have demonstrated that prenatal methadone exposure can lead to increased premature birth (Cleary et al, 2012; Fajemirokun-Odudeyi et al, 2006; Lejeune et al, 2006), decreased birthweight (Cleary et al, 2012; Hulse et al, 1997; Kandall et al, 1976; Sarfi et al, 2009; van Baar et al, 1994), and smaller head circumference (Brown et al, 1998; Hans, 1989; Rosen and Johnson, 1985; van Baar et al, 1994; Welle-Strand et al, 2013; Wilson et al, 1981). Additional reports indicate an increased incidence of respiratory insufficiency at birth (Maas et al, 1990), altered corrected QT interval on electrocardiogram during the first postnatal week (Parikh et al, 2011), postnatal hyperphagia (Martinez et al, 1999), disrupted auditory event related potentials (Paul et al, 2014), and myelination deficits (Walhovd et al, 2012). However, the prevalence of cognitive impairments produced by prenatal methadone has been questioned because some studies have not observed differences in cognitive development (de Cubas and Field, 1993; Hans, 1989; Rosen and Johnson, 1985) or performance (Bauman and Levine, 1986; Soepatmi, 1994; van Baar, 1990; van Baar and de Graaff, 1994; van Baar et al, 1994). These variations may be in part due to socioeconomic status and other variables (Hans, 1989).
In clinical studies of opiate maintenance therapies, we are aware of the prenatal opiate exposure dose, which allows researchers to mimic physiologically relevant plasma concentrations of the drug. Most animal studies do not measure plasma concentrations, but variations in dose and method of delivery are common issues in prenatal exposure studies. Despite these differences, fairly consistent alterations have been seen with prenatal methadone exposure in animals. Similar to models of perinatal heroin and morphine exposure, perinatal methadone exposure leads to decreased birthweight (Enters et al, 1991; Ford and Rhines, 1979; McLaughlin et al, 1978; Zagon and McLaughlin, 1977a; 1983, ), mu opioid receptor binding (Hou et al, 2004), hyperactivity (Hutchings et al, 1992) and reduced performance on learning and memory tasks (Vargas et al, 1975; Zagon and McLaughlin, 1979; Zagon et al, 1979). Decreased brain and cerebellar weight and brain DNA content (Ford and Rhines, 1979; McLaughlin et al, 1978; Zagon and McLaughlin, 1977a; 1983, ) as well as cortical thickness (Ford and Rhines, 1979) have also been seen. Zagon and McLaughlin (1982) found reduced cerebellar area and decreased number and density of internal granular neurons in the cerebellum with perinatal methadone exposure. A delay of developmentally timed behaviors was reported by the same group (Zagon and McLaughlin, 1978). Consistent with the cardiac abnormalities in neonates by Parikh and colleagues, rodent perinatal methadone exposure delays cellular development of the heart evident by altered levels of polyamines (Slotkin et al, 1982).
Alterations in the levels and/or activity of multiple neurotransmitter systems occur with perinatal methadone exposure. Studies have shown decreased hippocampal NE (Robinson et al, 1997), and disrupted striatal cholinergic activity compared with controls (Guo et al, 1990; Robinson, 2002; Robinson et al, 1996; Robinson and Wallace, 2001). McGinty and Ford found decreased DA in the forebrain at P1 and P20 in methadone-exposed rodents (McGinty and Ford, 1980). Prenatal only methadone exposure may decrease striatal DA turnover whereas combined pre- and postnatal exposure appears to increase it (Robinson et al, 1996). However, postnatal methadone exposure was required to decrease DA turnover in the frontal cortex (Robinson et al, 1997), which paints a complex picture of altered DA neurotransmission in perinatal methadone exposure. The 5-HT system shows irregularities in the transport system in the cortex and hippocampus (De Montis et al, 1983) and increased basal levels in the parietal cortex with perinatal methadone (Robinson et al, 1997). Additional disruptions in monoamine oxidase B levels (Tsang et al, 1986), METH sensitization (Wong et al, 2014), and startle response (Hutchings et al, 1993; Zmitrovich et al, 1994) also have implications for cognition and addiction vulnerability.
Buprenorphine
Buprenorphine is a partial mu opioid receptor agonist and a kappa opioid receptor antagonist that is given as an outpatient opioid maintenance therapy. Buprenorphine is not currently the recommended prenatal opioid maintenance therapy in the United States, although it is widely used elsewhere. A recent review of double-blind randomized control trials found insufficient evidence to recommend buprenorphine over methadone during pregnancy (Minozzi et al, 2013). This recommendation was largely based on a slightly superior retention of pregnant women in maintenance therapy with methadone, although buprenorphine may produce less severe neonatal abstinence syndrome (Minozzi et al, 2013). The American College of Obstetricians and Gynecologists has urged that buprenorphine be considered first-line treatment, but methadone is likely still the gold standard due to slightly higher adherence, more tightly controlled dosing, and insufficient evidence that buprenorphine is superior than methadone treatment (ACOG Committee, 2012). Observation of clinical cohorts has suggested that prenatal buprenorphine may produce fewer neurobehavioral problems (Coyle et al, 2012), higher birthweight, and larger head circumference compared with methadone (Welle-Strand et al, 2013). Several studies have found no difference in growth patterns between buprenorphine-exposed and non-exposed neonates (Bakstad et al, 2009; Fischer et al, 2000; Schindler et al, 2003; Sundelin Wahlsten and Sarman, 2013). Yet, hyperactivity, visual/motor impairment, and memory problems (Sundelin Wahlsten and Sarman, 2013) as well as an increase in premature birth compared with non-exposed neonates have been observed. Therefore, although there are still developmental perturbations observed with prenatal buprenorphine, the risk to the fetus may be less than with prenatal methadone, and we encourage physicians to consider buprenorphine as a first-line maintenance therapy with pregnant patients.
Fewer animal studies have been published for perinatal buprenorphine than methadone and heroin/morphine, but perinatal buprenorphine appears to produce alterations common to opiate exposure. Like other opiates, perinatal buprenorphine produces morphine tolerance (Chiang et al, 2010; Robinson and Wallace, 2001), delayed acquisition of developmentally timed behaviors (Robinson and Wallace, 2001), and increased sensitization to METH (Chiang et al, 2013). Chiang et al (2013) also found decreased D1 receptor mRNA, basal cyclic AMP, and D1 receptor induced adenylyl cyclase activity in the NAc of buprenorphine-exposed offspring, suggesting disrupted signal transduction may underlie the increased METH sensitization. Although mu opioid receptor binding is decreased at birth, it corrects by postnatal day 7 (Belcheva et al, 1998; Belcheva et al, 1994; Hou et al, 2004). Perinatal buprenorphine does not appear to cause hyperactivity in animal models to date, although this has been reported in some clinical studies (Hutchings et al, 1996; Sundelin Wahlsten and Sarman, 2013). Effects on striatal acetylcholine levels are dose dependent with high doses causing a decrease at P4 and 21 but low doses producing an increase at P21 (Guo et al, 1990; Robinson, 2002). Altered myelination, which has been observed in clinical studies of opiate-exposed neonates (Walhovd et al, 2012), also occurs, producing increased myelinated axon caliber with disproportionately thin myelin sheaths potentially due to changes in myelin basic proteins and myelin-associated glycosylation (Sanchez et al, 2008). Additional observations in animal models of perinatal buprenorphine exposure have been reviewed comprehensively by others (Farid et al, 2008).
Naltrexone
Naltrexone is a long-acting, nonselective opioid receptor antagonist used more commonly outside of the United States to prevent relapse in opiate addicts due to its ability to block the euphoric effects of opioid agonists, low tolerance and abuse potential, and modest adverse effects (Jones et al, 2013). Newly developed depot formulations allow for dosing every 4–6 weeks, improving patient adherence and making this an increasingly preferred treatment option. Inadvertent exposure due to conception after naltrexone depot implant placement in Australia, Portugal, and the United Kingdom has been cited, showing unremarkable neonatal and obstetric features compared with national averages as well as improved outcomes relative to methadone maintenance during pregnancy (Hulse and O’Neil, 2002; Hulse et al, 2003; Hulse et al, 2004; Hulse et al, 2001). However, these studies did not address the complications of pain management during labor and delivery or effects on postnatal development.
Animal studies suggest that caution should be taken with naltrexone exposure during pregnancy. Endogenous opioid growth factor and the zeta receptor (this was subsequently renamed the ‘opioid growth factor receptor’) are found in both rodents and humans developmentally and decrease cell proliferation (Zagon et al, 1992; Zagon et al, 1999). At supratherapeutic dosing, animal models show increased birth weight and brain and cerebellar weight likely through attenuation of endogenous opioid growth inhibition with no remarkable effects on gestation (McLaughlin et al, 1997a, 1997b; Zagon and McLaughlin, 1984; Zagon et al, 1998). Zagon et al (1998) supported this hypothesis by showing decreased cerebellar proliferation with perinatal naltrexone (Zagon and McLaughlin, 1987). Perinatal naltrexone also alters opioid receptor expression and function (Bardo et al, 1982; 1983, ; Medina Jimenez et al, 1997). High perinatal doses (50 mg/kg) also increase dendritic arbor and spine growth in the cerebellum and hippocampus (Hauser et al, 1987; 1989, ). In contrast, low doses of perinatal naltrexone appear to produce decreases in dendritic length, proliferation, and overall growth (Farid et al, 2012; Hauser et al, 1989; Zagon and McLaughlin, 1984, 1987). Although these data advocate that naltrexone exposure during pregnancy may not be harmless, dosing discrepancies with human studies confound interpretations. Postnatal human studies with accurate measurements of dose and timing of exposures along with neurobehavioral endpoints are needed.
Prenatal exposure to opiates thus causes long-lasting alterations in growth, cognition, and motor and visual abilities. Although evidence suggests that any treatment for opiate addiction improves maternal and fetal outcomes, each form of medical treatment brings their own risks. The gold standard, methadone, has good patient retention but causes a more pronounced neonatal abstinence syndrome whereas buprenorphine has higher abuse potential but causes a shorter, less severe neonatal abstinence syndrome. Both of these mu opioid receptor agonists may alter developmental trajectory and negatively impact cognition. The increasing use of opioid antagonist (naltrexone) depot formulations to treat opioid addiction will bring more cases of fetal exposure to naltrexone and an increased risk of opioid overdose and relapse during pregnancy.
CANNABIS/delta-9-tetrahydrocannabinol
Cannabis, also known as marijuana or, in its more concentrated form, hashish, is an illicit drug derived from the Cannabis sativa plant and is commonly smoked or, less frequently, ingested orally. For the purposes of this review, we will focus on the more commonly used form, marijuana, in its most popular route of administration, smoking. Marijuana is the most commonly abused form of illicit drug in the US, with over 42% of the population aged 12 years and over trying the drug within their lifetime. Over 38% of US females aged 12 and over will try the drug at some point in their lives, with 5% of females using the drug within the past month (Substance Abuse and Mental Health Services Administration, 2013). These statistics include women of childbearing age, and nearly 5% of pregnant women report smoking marijuana within the first trimester of pregnancy, although these numbers decrease in the second (2.9%) and third trimesters (1.4%) (Substance Abuse and Mental Health Services Administration, 2009). Given that cannabis has been recently legalized in Washington and Colorado and decriminalized in other states, with many others likely to follow suit, prenatal exposure to it is of growing concern.
Marijuana elicits a sedative-like effect, primarily due to its active ingredient, delta-9-tetrahydrocannabinol (THC). THC binds to the cannabinoid-1 (CB-1) receptor to induce this effect (for review, see Trezza et al (2008b)). Transcript for the CB-1 receptor is present in the second embryonic week in rodents (Buckley et al, 1998) and in the second trimester in humans (Wang et al, 2003), indicating that exposure to marijuana in the womb might have developmental consequences. Furthermore, THC can cross the placenta with reasonable efficiency (Hutchings et al, 1989), although unlike many other drugs of abuse, the placenta appears to limit fetal exposure to marijuana, as fetal THC concentrations have been documented to be lower than maternal concentrations in studies of various animal species (Behnke and Smith, 2013). Endogenous cannabinoids (endocannabinoids) are crucial for proper development in utero, and exogenous cannabinoids such as marijuana alter fetal growth trajectories (El Marroun et al, 2009), an effect that can have long-term consequences (for review, see Keimpema et al (2011)). Marijuana users can become dependent on the drug. Moreover, the THC content of marijuana has greatly increased over the years (Mehmedic et al, 2010), causing additional concern for fetal exposure.
Despite the overwhelming use among pregnant women, there are relatively few clinical studies looking at the effects of marijuana on the offspring. Studies have shown that fetuses exposed to marijuana during gestation exhibited stunted growth outcomes, as assessed by fetal weight and foot length (Hurd et al, 2005). Shorter gestation lengths, decreased birth weights, and deficits in other growth measures have been reported (Cornelius et al, 1995; Fergusson et al, 2002; Fried et al, 1999; Fried et al, 1984; Linn et al, 1983), although others have shown little to no effect on birth outcomes (Day et al, 1991; Witter and Niebyl, 1990). While the gestational and newborn growth outcomes remain equivocal, postnatal neurobehavioral outcomes are only slightly less so. Our understanding of the role of prenatal marijuana exposure derives primarily from two long-term longitudinal studies: the Ottawa Prenatal Prospective Study (OPPS) and the Maternal Health Practices and Child Development Study (MHPCD). The OPPS study, initiated in 1978, consists of primarily ‘low-risk’ middle class subjects, while the MHPCD, begun in 1982 in Pittsburgh, focuses on ‘higher risk’ subjects of lower socioeconomic status. As outlined below, evidence from these studies and others suggests that prenatal marijuana exposure has deleterious effects on exposed children in particular domains, although these results are not definitive (for review, see Fried and Smith (2001) and (Campolongo et al (2009)).
Newborns exposed to marijuana exhibit sleep disturbances (Scher et al, 1988), a problem that persists through age 3 (Dahl et al, 1995), as well as a shorter, high-pitched cry (Lester and Dreher, 1989). Altered responses to visual stimuli and increased startles and tremors were noted in newborns exposed to marijuana (Fried, 1982; Fried and Makin, 1987; Fried et al, 1987), with some such symptoms lasting for at least 30 days after birth (Fried et al, 1987), although others, also using the Brazelton Neonatal Behavioral Assessment Scale, have shown no significant differences on these outcomes (Tennes et al, 1985). Differential results were also seen in the MHPCD and OPPS cohorts using the Bayley Scales of Infant Development. Decreased mental scores in marijuana-exposed children at 9 months of age were found in the Pittsburgh group, an effect attributed to exposure during the third trimester, although at 19 months, no significant differences were seen relative to controls (Richardson et al, 1995). Meanwhile, others have shown no significant cognitive deficits in children ages 1–3 years that were prenatally exposed to marijuana (Astley and Little, 1990; Fried and Watkinson, 1988; 1990, ). However, exposed children at 3 years of age from the higher risk MHPCD study demonstrated attenuated cognitive development, including decreased short-term memory, verbal, and visual skills (Day et al, 1994). These effects correlated with exposure during the first and second trimester yet that can in some populations be ameliorated by environmental enrichment (Day et al, 1994).
As children approach school age, additional detrimental effects of prenatal marijuana exposure become apparent. These deficits are not generalized to overall cognition but are specific to higher order function, specifically, executive function. Impairments in verbal and memory tasks become apparent in children at age 4, despite exhibiting no such alterations when tested at younger ages (Fried and Watkinson, 1990). At 5–6 years of age, exposed children showed no overall deficits in cognition or language skills (Fried et al, 1992a), but 6–year-olds showed attention deficits, elevated impulsivity, and hyperactivity (Fried et al, 1992b; Leech et al, 1999). Deficits in short-term memory and verbal reasoning were apparent at this age as well (Goldschmidt et al, 2008). Over time, attention deficits remain and can escalate to increased delinquency and externalizing behaviors (Goldschmidt et al, 2000). Interestingly, a study on marijuana-exposed fetuses found elevated levels of D2 receptor transcript in the amygdala of males (Wang et al, 2004), suggesting potential for altered emotional regulation. Alterations in visuospatial memory are also noted in early adolescence (Fried and Smith, 2001). Deficits in these more complex domains were seen at ages 9–12 (Fried and Watkinson, 2000; Fried et al, 1998), although other data indicate that deficits in visual and abstract reasoning can be detected as early as 3 years of age (Griffith et al, 1994). Perhaps most striking is that even in their early 20 s, exposed individuals still have deficits in visuospatial working memory and impulsivity (Smith et al, 2004, 2006a). Adolescents performed more poorly in some areas of academic testing (Fried and Smith, 2001; Fried et al, 2003; Goldschmidt et al, 2012). These data indicate that prenatal marijuana exposure has significant effects on multiple neurobehavioral outcomes—deficits that are enduring, particularly at the level of executive function.
Exposed children also exhibit signs of neuropsychiatric disorders and may be more susceptible to substance abuse (for review, see Jutras-Aswad et al (2009)). For example, children from the MHPCD cohort presented with signs of depression as early as 10 years of age (Gray et al, 2005). Moreover, decreased levels of D2 receptor transcript were found in the NAc of exposed fetuses, potentially linking susceptibility to drug use to prenatal marijuana exposure (DiNieri et al, 2011). Prenatally exposed children were also more likely to experiment with marijuana at an earlier age as well as smoke the drug more frequently (Day et al, 2006; Goldschmidt et al, 2012). Given that drug use and psychiatric disorders are burdens not only to the individual but also to society as a whole, in total these results indicate the prenatal marijuana exposure has persistent deleterious effects that can have considerable consequences.
Animal models of prenatal marijuana exposure support these findings. Exposed offspring showed reduced body weight at birth (Abel et al, 1981; Abel et al, 1980). As with humans, young rats exposed to THC throughout pre- and postnatal development had increased ultrasonic vocalizations (cries) when isolated, indicative of increased anxiety and potentially analogous to increased irritability in human infants (Trezza et al, 2008a). In this same study, rats showed deficits in social behaviors during adolescence and elevated anxiety in adulthood. Increased locomotor activity has been demonstrated as well (Borgen et al, 1971; Mereu et al, 2003; Rubio et al, 1995), although others have shown either no changes in locomotion or significant hypoactivity (for review, see Navarro et al (1995); Schneider (2009)).
As previously mentioned, marijuana exposure during human prenatal development appears to affect ‘higher order’ facets of learning and memory. Similar results have been found in animal models as well. Using either the CB-1 receptor agonist WIN55,212-2 (Mereu et al, 2003) or THC (Campolongo et al, 2007), researchers found that developmental exposure resulted in impairments in passive avoidance, indicative of diminished memory retention. Furthermore, the THC-treated group also was unable to discriminate between a novel and a familiar rat in the social discrimination test. These effects are not limited to a particular phase of life, as in utero-exposed rats showed deficits in learning and memory tasks throughout the lifespan (Silva et al, 2012). These data and others (Campolongo et al, 2009; Schneider, 2009) indicate that marijuana exposure during pregnancy impacts learning and memory. Marijuana exposure during development may also result in increased likelihood of drug experimentation later in life. Exposed rats exhibited decreased levels of D2 receptor expression and binding in the NAc, a key reward center, and increased sensitivity to the rewarding properties of opiates (DiNieri et al, 2011). Others have shown that exposure to THC during a period equivalent to mid-gestation in humans resulted in increased heroin-seeking behavior in offspring (Spano et al, 2007). Following a similar exposure paradigm, rats administered AMPH in adulthood were less sensitive to its locomotor-stimulating effects (Silva et al, 2012), indicative of aberrant responsiveness to psychostimulants. While the preclinical data support the clinical findings, further research is needed to understand the mechanism and circuitry underlying these deficits.
TOBACCO/NICOTINE
According to the 2010 Pregnancy Risk Assessment Monitoring System data from the CDC, 12.3% of pregnant women in the US continue to smoke throughout pregnancy (Tong et al, 2013), and these numbers might be higher worldwide (Bloch et al, 2008). Over 7000 different chemicals are found in cigarette smoke, many of which are toxic and/or carcinogenic (U.S. Department of Health and Human Services, 2010). Nicotine, the primary psychoactive component of tobacco, and its metabolite, cotinine, readily pass through the placenta, and fetal concentrations of these compounds are significantly greater than those achieved by the mother (Lambers and Clark, 1996). Nicotine is an agonist of the nicotinic acetylcholine receptor (nAChR), which is expressed in the first trimester in humans and by the second gestational week in rodents (see review Dwyer et al (2008); Dwyer et al (2009)). Acting through this receptor, developmental exposure to nicotine disrupts the cholinergic system, a key modulator of brain development, and alters synaptogenesis, neuronal migration, neurotransmitter release, and a host of other molecular and functional endpoints (see review of Slotkin 2004). However, nicotine is only 1 of more than 4000 compounds to which the fetus is exposed through maternal smoking (Behnke and Smith, 2013).
Maternal smoking is not the only exposure to nicotine for the fetus. Maternal exposure to secondhand smoke (SHS) is also highly detrimental to proper fetal development and can cause long-term neurobehavioral alterations on its own (Chen et al, 2013). In addition, nicotine replacement therapies (ie, lozenges, patches, gum, etc.) are prescribed more and more to reduce fetal exposure to the additional compounds found in cigarette smoke (Coleman, 2008). However, nicotine alone, based on animal studies, is highly toxic in and of itself, and these devices should not be considered ‘safe’ for pregnant women to use, although the risks are significantly less compared with cigarette smoking alone (for review, see Bruin et al (2010)). Along those same lines, electronic cigarettes (e-cigarettes) have become increasingly popular as they eliminate SHS by delivering nicotine as an inhalable vapor. These devices should be used with caution, as they expose not only the user, but also passerby to aerosolized nicotine (Czogala et al, 2013). Similarly, maternal use of smokeless (chewing) tobacco not only decreases birth weight (England et al, 2003), a risk factor for long-term neurobehavioral consequences (Rosenthal et al, 2011), but also results in higher Lipsitz scores, a neurobehavioral test used to measure drug withdrawal symptoms such as tremor, increased muscle tone and reflexes, and irritability in newborns (Hurt et al, 2005).
There is a wealth of data examining the effects of prenatal exposure to tobacco in both humans and animals, and a number of very detailed reviews exist on the consequences of prenatal tobacco exposure (Abbott and Winzer-Serhan, 2012; Bublitz and Stroud, 2012; Cornelius and Day, 2009; Pauly and Slotkin, 2008; Slotkin, 2008). For this review, we will provide an overview of some of the findings pertaining to neurobehavioral outcomes. At birth, exposed infants tend to be smaller in body weight, height, and head circumference, effects attributable to third and possibly second trimester exposure (Espy et al, 2011; Himes et al, 2013). There is also a greater likelihood of exposed infants to be admitted into the neonatal intensive care unit relative to unexposed controls (Adams et al, 2002). Newborns exposed in utero to nicotine are more irritable and have poorer attention than unexposed infants, and they exhibit hypertonicity, increased tremors and startle responses, and deficient speech processing as well (Espy et al, 2011; Fried et al, 1987; Key et al, 2007; Mansi et al, 2007; Reijneveld et al, 2002; Stroud et al, 2009a). Within the first month of life, exposed infants show signs of poorer self-regulation and require more handling by caregivers (Stroud et al, 2009b). Newborns also demonstrate an attenuated response to auditory stimuli, an effect that can contribute to language and learning impairments later in life (Kable et al, 2009; Mansi et al, 2007).
Following birth, the consequences of prenatal nicotine exposure are persistent, with some outcomes being consistently shown by multiple independent reports. Children age 6 and younger had decreased receptive language skills (Fried et al, 1992a; Fried and Watkinson, 1990; Lewis et al, 2007), which can contribute to language comprehension deficits. Exposed children also show poorer academic achievement or cognitive scores than peers (Agrawal et al, 2010; Fried et al, 1992a; Fried and Watkinson, 1990). These effects are not limited to young children. Older adolescents also exhibit cognitive deficits, as 16–18 year olds, exposed to tobacco in utero and were current smokers themselves, showed deficits in sensory processing (ie, visuospatial memory) when abstinent from nicotine (Jacobsen et al, 2006). Interestingly, this effect was absent when subjects were allowed to smoke freely, indicating that the deficit was withdrawal-induced and that smoking was a means to compensate for this deficit. Furthermore, there was no difference between subjects exposed to tobacco throughout pregnancy or during only the first trimester, emphasizing the necessity of smoking cessation before pregnancy. In a test of working memory (N-back test) combined with fMRI, there was differential activation of various brain regions following correct responses in exposed versus unexposed adolescents, indicating abnormal function or circuitry following exposure (Bennett et al, 2013). Moreover, pallidum volume was smaller in adolescents following prenatal tobacco exposure, and increased impulsivity was correlated with thalamic volume following prenatal tobacco (Liu et al, 2013).
Attention is also a key area where a great number of deficits are seen following gestational tobacco exposure. Studies have shown that children as young as 6 years old show attention deficits, including diagnosis with ADHD, an effect seen well into the late teens (Bennett et al, 2009; Cornelius et al, 2011; Cornelius et al, 2007; Jacobsen et al, 2007; Kotimaa et al, 2003; Langley et al, 2007; Lindblad and Hjern, 2010). Both second (Cornelius et al, 2007) and third (Cornelius et al, 2011) trimester exposures have been correlated to attention problems. Interestingly, individuals with self-reported ADHD or attention problems were more likely to smoke than their peers (Kollins et al, 2005), further corroborating a link between attention disorders and nicotine use. While gestational exposure to nicotine appears to be a primary indicator for attention deficits, confounding factors such as maternal diagnosis of ADHD, environmental factors, socioeconomic status, and exposure to other drugs are often not accounted for in these studies (Agrawal et al, 2010; Ball et al, 2010; Langley et al, 2012). Thus, more thorough research to eliminate these factors as confounding variables is necessary.
Conduct and behavioral disorders are other domains frequently associated with prenatal tobacco exposure. Children as young as 18–24 months show increases in externalizing behaviors (Stene-Larsen et al, 2009; Wakschlag et al, 2006a) as well as internalizing and total problem scores following prenatal tobacco exposure (Carter et al, 2008). A study of 3-year-olds exposed gestationally to tobacco determined that increases in oppositional behavior were correlated to third trimester exposure (Day et al, 2000). Similar behavioral issues, including aggression, oppositional defiance, and delinquency, have been noted in older children (6–16 years old) (Bennett et al, 2009; Cornelius et al, 2011; Cornelius et al, 2007; Indredavik et al, 2007; Langley et al, 2007; Wakschlag et al, 2006b; Weitzman et al, 1992). It would appear that no level of tobacco exposure may be safe, as children aged 7–15 years, born of non-smoking mothers with SHS exposure during pregnancy, showed increased externalizing behavior (Gatzke-Kopp and Beauchaine, 2007). Moreover, children with only prenatal exposure to tobacco were more likely to exhibit abnormal behaviors relative to children exposed only during the postnatal period (Ruckinger et al, 2010), thus emphasizing that gestation is a critical period in the development of behavioral disorders.
In addition to the aforementioned deficits, prenatally exposed children are more likely to develop substance use disorders. Lotfipour et al (2009) found that exposed children were more likely to experiment with drugs, an effect correlated to thinning of the orbitofrontal cortex. This same group also demonstrated that prenatal tobacco exposure, in combination with a genetic polymorphism of the α6 subunit of the nAChR, resulted in increased likelihood of smoking and drug use (Lotfipour et al, 2010). The combined environmental and genetic conditions altered brain development as well, as affected children had larger striatal volumes. Others have shown that exposed children begin to smoke and become regular users earlier than unexposed peers (Agrawal et al, 2010).
While preclinical models allow for more specific and controlled studies, variables such as species differences, pharmacokinetics, routes of administration, and the timing of the developmental exposure make comparisons or conclusions difficult. These studies, however, are invaluable in defining critical periods and insight into the underlying molecular mechanisms and circuitry following developmental nicotine exposure. For instance, tobacco smoke exposure in non-human primates during gestation and lactation resulted in neuronal cell loss, increased glia, and decreases in cell size (Slotkin et al, 2006). Following nicotine exposure in utero, changes to spine density, dendritic length, and dendritic branching were observed in juvenile (Muhammad et al, 2012) and adult (Mychasiuk et al, 2013) rats. Such changes can alter the trajectory of development and contribute to the functional deficits seen later in life (Pauly and Slotkin, 2008).
Changes in body weight are also found in animal models of developmental nicotine exposure. When pregnant dams were exposed to cigarette smoke throughout gestation, fetuses were significantly smaller than controls (Bassi et al, 1984; Esposito et al, 2008). Differences in body weight following other routes of administration or only postnatal exposure (equivalent to third trimester human exposure, see Figure 1) have been documented as well, although results are variable (see (Abbott and Winzer-Serhan (2012)) for review). In line with the clinical data, deficits in learning and memory and sensory processing have also been detected. Rodents exposed in utero to nicotine performed poorly on tests of learning and memory, including the radial arm maze (Levin et al, 1993; Sorenson et al, 1991) and two-way active avoidance (Vaglenova et al, 2008). Similarly, mice exposed to nicotine via injection took longer to reach criterion on the radial arm maze and had increased latencies to reach the platform on the Morris water maze (Yanai et al, 1992). Exposed mice exhibited deficits in sensory processing, as evidenced by hypersensitive passive avoidance (Heath et al, 2010). These researchers determined that postnatal exposure—the equivalent of the human third trimester—was the key critical period for this effect. A study using a mouse model of inhaled cigarette smoke throughout gestation through weaning (simulating exposure throughout human pregnancy) resulted in deficits in spatial and reference memory as assessed via Morris water maze but not in route-based (egocentric) learning in the Cincinnati water maze (Amos-Kroohs et al, 2013). These findings bolster the clinical evidence that prenatal nicotine exposure alters long-term learning and memory function.
Clinical data on developmental nicotine exposure frequently refers to hyperactivity and attention deficits, effects that are found in the preclinical literature as well. Schneider et al (2011) found that rats exposed to nicotine during gestation performed more poorly in the 5-choice serial reaction time task in measures of sustained attention and impulsivity (Schneider et al, 2011). These data correspond to the clinical literature, specifically as attention deficits have been correlated to second trimester exposure (Cornelius et al, 2007). A similar exposure regimen in mice found that males and females exposed to nicotine were significantly more hyperactive than controls, especially during the dark (awake) cycle (Zhu et al, 2012). Moreover, nicotine-exposed mice responded to methylphenidate (MPH), a pharmacological treatment for ADHD, with attenuated locomotor activity. This low oral dose of MPH had no effect on control mice, just as humans without ADHD do not respond to MPH. Perhaps even more amazingly, the authors then demonstrated that the prenatal nicotine-induced hyperactivity can propagate transgenerationally via the maternal line of descent (Zhu et al, 2014). These data suggest that in utero exposure to substances even in previous generations may have a role in the increased incidence of neurodevelopmental disorders in modern society.
Finally, while conduct disorders per se are difficult to measure in animal models, comorbid conditions, such as anxiety, can be modeled. In fact, evidence exists that prenatal nicotine increases anxiety in rodents (Huang et al, 2007; Santiago and Huffman, 2013; Vaglenova et al, 2008), although also see Amos-Kroohs et al (2013). Exposed animals are more likely to self-administer nicotine later in life (Chistyakov et al, 2010; Levin et al, 2006). Others have shown that exposed rodents are more sensitive to the rewarding (Harrod et al, 2012) and locomotor-stimulating (Amos-Kroohs et al, 2013) properties of METH. Based on these data, further research into the long-term alterations resulting from developmental nicotine exposure, specifically into aggression, abnormal social behavior, and substance use disorders, is needed.
ALCOHOL
Alcohol consumption during pregnancy can result in a long-recognized fetal alcohol syndrome (FAS), which includes morphogenic effects on limb and facial development, reduced brain and birth weight, and cognitive delays and impairments (Behnke and Smith, 2013; Foltran et al, 2011; Jones, 1986). However, even moderate exposure can impact brain development and these effects persist at least into young adulthood. While FAS represents the most severe manifestation of heavy maternal alcohol consumption during pregnancy, the term ‘fetal alcohol spectrum disorders’ (FASDs) has been applied to characterize a broad range of deficits present in individuals with or without facial dysmorphology who were exposed to alcohol prenatally (Foltran et al, 2011; Hoyme et al, 2005; Nash et al, 2008; Riley and McGee, 2005). Thus, moderate alcohol exposure during fetal development produces significant neurobehavioral consequences (Behnke and Smith, 2013; Day and Richardson, 1991; 2004, ; Fried and Watkinson, 1988; Jacobson and Jacobson, 1999; Jacobson et al, 1993; Nash et al, 2013), and FASDs currently represent the leading cause of mental retardation in North America, ahead of Down syndrome and cerebral palsy (Nash et al, 2008). As described in a 1996 Institute of Medicine Report to Congress—‘Of all the substances of abuse (including cocaine, heroin, and marijuana), alcohol produces by far the most serious neurobehavioral effects in the fetus.’ (Institute of Medicine, 1996). It has been estimated that at least 1 per 100 (1% of live births) are affected by FAS or FASDs (Sampson et al, 1997). Twelve percent of pregnant women self-report alcohol use within the past month (Substance Abuse and Mental Health Services Administration, 2008). A recent Australian study found that women who binged only before pregnancy were more likely to continue (55%) rather than reduce drinking (29%) once pregnant (Anderson et al, 2014). In 2002, the estimated societal cost for each individual with FAS was $2.0 million ($1.6 million for medical treatment, special education, and residential care for persons with mental retardation, and $0.4 million for lost productivity) (Lupton et al, 2004). The cost of FASD in the United States was last estimated at $4–8 billion annually (Popova et al, 2011). For comparison, the annual budget of the entire National Institute on Alcohol Abuse and Alcoholism is substantially less than 500 million.
In addition to total quantity, patterns of alcohol drinking can also modulate the severity of resultant deficits—for example ‘moderate’ drinking has much more impact on child development when the mother consumes several drinks in a single day than when she drinks the same quantity in doses of one to two drinks per-day over several days (Jacobson and Jacobson, 1999). Imaging studies reveal gray matter volume in the left cingulate gyrus, bilateral middle frontal gyri, right middle temporal gyrus, and right caudate nucleus following low-to-moderate exposure (De Guio et al, 2014; Eckstrand et al, 2012; Treit et al, 2013). Fetal alcohol has also been associated with a host of cognitive and behavioral impairments, including deficits in attention, memory, verbal fluency, executive functioning, reaction time, and motor learning (Coles et al, 2002; Howell et al, 2006; Mattson et al, 1998; Richardson et al, 2002; Riley and McGee, 2005; Willford et al, 2004). Maternal and fetal alcohol dehydrogenase polymorphisms also modulate risk (Warren and Li, 2005).
Ethanol has diverse cellular targets, and produces wide-ranging effects depending on the dose, duration, and timing of exposure. Current evidence suggests that alcohol produces many of its damaging effects by exerting specific actions on molecules that regulate key developmental processes (eg, L1 cell adhesion molecule, alcohol dehydrogenase, catalase), interfering with trophic factors that regulate neurogenesis and cell survival, and/or inducing excessive cell death via oxidative stress or activation of caspase-3 proteases (Conti et al, 2009; Farber et al, 2010; Goodlett et al, 2005; Ungerer et al, 2013). Several studies have identified effects of low amounts of alcohol exposure on the development of serotonergic neurons (Sari and Zhou, 2004; Zhou et al, 2003; Zhou et al, 2005), and suggested that these types of defects in the ontogeny of a specific neurotransmitter system may underlie the more ‘subtle’ deficits observed in human children diagnose with Alcohol Related Neurodevelopmental Disorder, rather than full-blown FAS (Zhou et al, 2003). Ethanol exposure disrupts the proliferation of glia and neuronal precursors in the developing central nervous system (Luo and Miller, 1998; Miller, 2007; Miller and Hu, 2009; Powrozek and Miller, 2009) and can dramatically alter behavior (Hellemans et al, 2010; Valenzuela et al, 2012). Transcriptional analyses of gene products regulated by fetal ethanol exposure in both mouse and human cells also highlight genes related to neural development, such as cell proliferation, neuronal migration, and differentiation (Hashimoto-Torii et al, 2011; Hicks et al, 2010). Emerging data also implicate microRNAs and epigenetic mechanisms (Balaraman et al, 2013; Mantha et al, 2014; Pappalardo-Carter et al, 2013; Ungerer et al, 2013). For example, fetal alcohol results in alterations in DNA methylation and microRNA expression and function (Pappalardo-Carter et al, 2013). An intriguing hypothesis suggests that fetal alcohol may induce a dual state of central nervous system insulin resistance and oxidative stress and proposes that adverse effects may be prevented or treated by treatment with peroxisome-proliferated activated receptor agonists (de la Monte and Wands, 2010).
Alcohol exposure continues to have potent effect on brain development postnatally in rodents, demonstrated by studies showing dramatic effects on the number of layer 2/3 cortical pyramidal neurons, on the branching patterns of their basilar dendrites (Granato et al, 2003; Granato and Van Pelt, 2003), and on dendritic excitability (Granato et al, 2012). The same group has gone on to also implicate distinct alterations in cerebral cortical and striatal interneuron distribution and morphology (De Giorgio et al, 2012; Granato, 2006). Rodent postnatal alcohol exposure also leads to a persistent disruption in ocular dominance plasticity, which can be treated, at least in part, with overexpression of serum response factor or inhibition of a phosphodiesterase (Medina et al, 2006; Paul et al, 2010). Once again, these time periods are most similar to late prenatal development in humans (Figure 1).
Finally, although we focus here on fetal exposures, of great additional interest is the suggestion that adolescents respond differentially to the acute cognitive, sensorimotor, and behavioral effects of alcohol consumption than adults (Spear, 2014; Spear and Varlinskaya, 2005). For example, studies from Brunell and Spear (2006) indicate that adolescents exhibit bimodal behavioral responses to alcohol, with both hypo- and hyperresponsive systems (Doremus et al, 2003; Long et al, 1992; Varlinskaya and Spear, 2002). These findings have important implications for the risks associated with recreational drinking in adolescents and young adults.
Taken together, alcohol exposure during development induces behavioral, cognitive, and neural alterations that are specific, replicable, persistent and highly dependent on the timing of the exposure. Given the staggering socioeconomic toll of prenatal alcohol exposures, new strategies to prevent exposures must be considered, and better mechanistic understandings are needed to facilitate potential ameliorative therapies.
CAFFEINE
There have been few studies about the effects of the naturally occurring adenosine receptor antagonist caffeine, which is ubiquitously consumed in coffee, tea, and cocoa-containing foods. Caffeine is likely the psychostimulant substance most consumed worldwide. Although considered a fairly innocuous drug, a recent animal study has provided compelling mechanistic data suggesting that developmental caffeine exposure transiently antagonizes adenosine receptor signaling resulting in discrete but persistent alterations in hippocampal structure and function (Silva et al, 2013). An accompanying editorial expressed concern that ‘We must now heed the wake-up call sounded by Silva et al (2013) that exposure to psychoactive substances, even a seemingly innocuous, socially well-integrated substance like caffeine, before or soon after birth can alter brain development’ (Kabir et al, 2013). Another team has suggested that embryonic caffeine exposure produces an increase in body weight (∼10%), alters cardiac function and morphology, and produces long-lasting alterations in DNA methylation (Buscariollo et al, 2014). A third group has focused on the neuroendocrine consequences and found that prenatal caffeine ingestion induces an increased susceptibility to metabolic syndrome with alterations of glucose and lipid metabolic phenotypes that then propagate transgenerationally (Katayama et al, 1987; Xu et al, 2012a; Xu et al, 2012b).
These recent studies join a larger body of work also suggestive of caffeine-induced changes in developmental trajectories (for review, see Porciuncula et al (2013)). Gressens et al (2001) have documented caffeine-induced acceleration of the evagination of neuroepithelium into telencephalic vesicles (Sahir et al, 2000) and upregulation of sonic hedgehog (Sahir et al, 2004). Gestational caffeine also downregulates central adenosine receptors (Leon et al, 2005; Lorenzo et al, 2010), reduces NMDA antagonist-induced locomotor activity (da Silva et al, 2005), and affects respiratory control (Bodineau et al, 2003; Saadani-Makki et al, 2004). Cognitive and motor deficits have also been reported in adult rodents exposed in caffeine during gestation and/or early postnatal life (Bjorklund et al, 2008; Soellner et al, 2009), although some impairments may be paradigm specific (Soellner et al, 2009).
The metabolism of caffeine and other methylxanthines is impaired in pregnant women, fetuses and neonates, leading to reduced clearance and potentially higher circulating levels (Aden, 2011). Several clinical studies have focused on concerns regarding caffeine-induced fetal growth restriction and have led to suggestions that women should reduce caffeine intake before conception and throughout pregnancy (Bracken et al, 2003; CARE Study Group, 2008). Other studies have failed to find an association (reviewed in Brent et al, 2011). Regarding more subtle changes in developmental trajectories, a recent study from the Netherlands did not find significant effects of prenatal caffeine on neurobehavioral functions at 5–6 years of age (Loomans et al, 2012), although data on caffeine intake was obtained only once during the ∼16th week of gestation. In contrast, a Norwegian prospective study has reported a small but significant association between maternal caffeine intake and inattention/overactivity based on the Child Behavior Check List (Bekkhus et al, 2010). Given the fairly dramatic range in caffeine metabolism rates among individual women (based on CYP1A expression and activity), it seems crucial that future studies employ pharmacogenomics strategies and caffeine metabolite measurements, rather than simple self-report of intake. And when combined with a still-evolving literature implicating adenosine A1 and A2A receptors in establishing neuronal homeostasis, tuning the ability of synapses to undergo plasticity, and in modulating neurotoxicity (Dias et al, 2013), these data suggest that expanded study of this topic is sorely needed.
FUTURE DIRECTIONS AND CLINICAL IMPLICATIONS
In summary, fetal exposure to drugs of abuse often produces long-lasting changes in brain structure and function (see Figure 3). These (mal)adaptations are regardless of whether the drugs are legal or illicit; it is in fact arguable that alcohol and nicotine produce more dramatic deficits than ‘harder’ drugs like cocaine. Adolescence encompasses another sensitive period of development; given that this is when illicit drug use typically begins, detailed research on how drugs of abuse alter relatively late-developing processes in the brain is also essential. Importantly, the molecular targets of psychoactive drugs sometimes have quite different actions in early development, as compared with their adult roles as synaptic modulators. In some cases, transgenerational effects via epigenetic changes have been documented. In addition, studies have repeatedly pointed out the crucial covariates of postnatal environment and socioeconomic status. The developing brain is extremely plastic and adaptable; we continue to discover new and better strategies for intervention.

Schematic summary of effects of distinct drug classes on offspring development. A wide variety of significant structural and neurobehavioral deficits are induced by fetal exposures to abused substances. As described in the text, the drug class, timing, dose, and pattern of intake all substantially determine the long-term effects on the developing child.
Surprisingly, research in this area is still in its formative stages, and more precise and mechanistic work will be necessary to characterize the extent of neurobehavioral alterations and how developmental timing, dosages, and genetics affect these processes. When costs for special education, long-term medical management, and lost productivity across the lifespan are considered, the socioeconomic impact is truly staggering. Moreover, societal responses continue to be misguided. For example, the state of Tennessee passed legislation in 2014 allowing women to be charged with assault if they have a pregnancy complication after using narcotics illegally. The desire to guard fetal health and outcomes is understandable; however, pregnant women who fear prosecution and the potential loss of their children will rarely seek essential prenatal and medical care. We already discovered the negative consequences of such practices during the peak of maternal cocaine exposures in South Carolina from 1989–1994, before their prosecution of these women was forced to cease (Annas, 2001; Budetti, 1993; Ferguson v. City of Charleston, 2001; McKnight v. State of South Carolina, 2008; Poland et al, 1993). Physicians and scientists need to better engage and educate the public on these complex issues.
We do not claim to have all the answers, or even all of the questions. But as a field, we must do a better job of bridging the translational gap. Basic scientists must design their animal models with consideration to (a) whether the route of administration (to the dam or the pup) is the same as in human users, (b) whether pharmacokinetics and biotransformation of the drug differ between humans and the animal model, (c) whether the model encompasses the entirety of fetal development, and (d) how well the outcome measures being tested in the animals conform to relevant outcome measures in the clinical literature. Given that women rarely, if ever, begin using an abused drug during the third trimester of their pregnancy, postnatal-only administration in rodents will usually be inadequate (Figure 1). Given the great prevalence of polysubstance use in drug-abusing women (and men), more models of prenatal polydrug exposure seem warranted. Interactions between fetal drug exposure and other environmental variables, including toxicants and stressors, are also crucial (Graf et al, 2013). Clinicians must also make better use of the preclinical findings and predictions about the kinds of deficits to be expected in people who have been exposed to substances prenatally (Figure 3). Particular focus should be paid when diverse types of models (species, dosing, timing, etc.) produce congruent effects on functional domains (even if the precise cellular mechanisms differ)—these are the maladaptations that will likely be most prevalent in the human population. The ability of animal data to potentially predict future health issues should also not be overlooked—for example, aging studies in animal models predict that prenatal exposure to cocaine or nicotine (Abreu-Villaca et al, 2004; Lawrence et al, 2008; Lloyd et al, 2006; Meyer et al, 2009) may produce higher risks or expression of cell death and/or neurodegenerative diseases later in life. While only time will tell, we will likely be facing additional societal and medical burdens as drug-exposed children continue to age—better animal models will allow us to predict and treat later-in-life disorders in these populations as they emerge.
FUNDING AND DISCLOSURE
We are currently supported by RO1MH086629 (GDS), R21DA035588 (GDS), the Vanderbilt Conte Center (GDS and EJR), the Peter F. McManus Charitable Trust (DLG), and the Vanderbilt Brain Institute. The authors declare no conflicts of interest.
References
Abar B, Lagasse LL, Wouldes T, Derauf C, Newman E, Shah R et al (2013). Cross-national comparison of prenatal methamphetamine exposure on infant and early child physical growth: a natural experiment. Prev Sci 1: 1–10.
Abbott LC, Winzer-Serhan UH (2012). Smoking during pregnancy: lessons learned from epidemiological studies and experimental studies using animal models. Crit Rev Toxicol 42: 279–303 Thorough review of the most recent literature regarding developmental tobacco exposure.
Abel EL, Bush R, Dintcheff BA, Ernst CA (1981). Critical periods for marihuana-induced intrauterine growth retardation in the rat. Neurobehav Toxicol Teratol 3: 351–354.
Abel EL, Dintcheff BA, Day N (1980). Effects of marihuana on pregnant rats and their offspring. Psychopharmacology (Berl) 71: 71–74.
Abreu-Villaca Y, Seidler FJ, Slotkin TA (2004). Does prenatal nicotine exposure sensitize the brain to nicotine-induced neurotoxicity in adolescence? Neuropsychopharmacology 29: 1440–1450.
ACOG Committee (2012). ACOG Committee Opinion No. 524: opioid abuse, dependence, and addiction in pregnancy. Obstet Gynecol 119: 1070–1076.
Adams EK, Miller VP, Ernst C, Nishimura BK, Melvin C, Merritt R (2002). Neonatal health care costs related to smoking during pregnancy. Health Econ 11: 193–206.
Aden U (2011). Methylxanthines during pregnancy and early postnatal life. Handb Exp Pharmacol 200: 373–389.
Agrawal A, Scherrer JF, Grant JD, Sartor CE, Pergadia ML, Duncan AE et al (2010). The effects of maternal smoking during pregnancy on offspring outcomes. Prev Med 50: 13–18.
Aguirre N, Barrionuevo M, Lasheras B, Del Rio J (1998). The role of dopaminergic systems in the perinatal sensitivity to 3, 4-methylenedioxymethamphetamine-induced neurotoxicity in rats. J Pharmacol Exp Ther 286: 1159–1165.
Amos-Kroohs RM, Williams MT, Braun AA, Graham DL, Webb CL, Birtles TS et al (2013). Neurobehavioral phenotype of C57BL/6J mice prenatally and neonatally exposed to cigarette smoke. Neurotoxicol Teratol 35: 34–45.
Anderson AE, Hure AJ, Forder PM, Powers J, Kay-Lambkin FJ, Loxton DJ (2014). Risky drinking patterns are being continued into pregnancy: a prospective cohort study. PLoS One 9: e86171. This sobering study found that over a third of women continue risky drinking into pregnancy, especially binge drinking. The authors suggest a need to address alcohol consumption prior to pregnancy.
Annas GJ (2001). Testing poor pregnant patients for cocaine—physicians as police investigators. N Engl J Med 344: 1729–1732.
Arjune D, Bodnar RJ (1989). Post-natal morphine differentially affects opiate and stress analgesia in adult rats. Psychopharmacology (Berl) 98: 512–517.
Aroyewun O, Barr GA (1982). The effects of opiate antagonists on milk intake of preweanling rats. Neuropharmacology 21: 757–762.
Aroyewun OO, Barr GA (1983). Effects of chronic antenatal and postnatal administration of narcotics on naloxone-induced anorexia in preweanling rats. Neuropharmacology 22: 329–336.
Arria AM, Derauf C, LaGasse LL, Grant P, Shah R, Smith L et al (2006). Methamphetamine and other substance use during pregnancy: preliminary estimates from the infant development, environment, and lifestyle (IDEAL) study. Matern Child Health J 10: 293–302.
Astley SJ, Little RE (1990). Maternal marijuana use during lactation and infant development at one year. Neurotoxicol Teratol 12: 161–168.
Bakstad B, Sarfi M, Welle-Strand GK, Ravndal E (2009). Opioid maintenance treatment during pregnancy: occurrence and severity of neonatal abstinence syndrome. A national prospective study. Eur Addict Res 15: 128–134.
Balaraman S, Tingling JD, Tsai PC, Miranda RC (2013). Dysregulation of microRNA expression and function contributes to the etiology of fetal alcohol spectrum disorders. Alcohol Res 35: 18–24.
Ball SW, Gilman SE, Mick E, Fitzmaurice G, Ganz ML, Seidman LJ et al (2010). Revisiting the association between maternal smoking during pregnancy and ADHD. J Psychiatr Res 44: 1058–1062.
Bandstra ES, Morrow CE, Accornero VH, Mansoor E, Xue L, Anthony JC (2011). Estimated effects of in utero cocaine exposure on language development through early adolescence. Neurotoxicol Teratol 33: 25–35.
Bardo MT, Bhatnagar RK, Gebhart GF (1982). Differential effects of chronic morphine and naloxone on opiate receptors, monoamines, and morphine-induced behaviors in preweanling rats. Brain Res 256: 139–147.
Bardo MT, Bhatnagar RK, Gebhart GF (1983). Age-related differences in the effect of chronic administration of naloxone on opiate binding in rat brain. Neuropharmacology 22: 453–461.
Barg J, Simantov R (1989). Developmental profile of kappa, mu and delta opioid receptors in the rat and guinea pig cerebellum. Dev Neurosci 11: 428–434.
Barr AM, Panenka WJ, MacEwan GW, Thornton AE, Lang DJ, Honer WG et al (2006). The need for speed: an update on methamphetamine addiction. J Psychiatry Neurosci 31: 301–313.
Bashore RA, Ketchum JS, Staisch KJ, Barrett CT, Zimmermann EG (1981). Heroin addiction and pregnancy. West J Med 134: 506–514.
Bassi JA, Rosso P, Moessinger AC, Blanc WA, James LS (1984). Fetal growth retardation due to maternal tobacco smoke exposure in the rat. Pediatr Res 18: 127–130.
Bauman PS, Levine SA (1986). The development of children of drug addicts. Int J Addict 21: 849–863.
Behnke M, Smith VC (2013). Prenatal substance abuse: short- and long-term effects on the exposed fetus. Pediatrics 131: e1009–e1024.
Bekkhus M, Skjothaug T, Nordhagen R, Borge AI (2010). Intrauterine exposure to caffeine and inattention/overactivity in children. Acta Paediatr 99: 925–928.
Belcheva MM, Bohn LM, Ho MT, Johnson FE, Yanai J, Barron S et al (1998). Brain opioid receptor adaptation and expression after prenatal exposure to buprenorphine. Brain Res Dev Brain Res 111: 35–42.
Belcheva MM, Dawn S, Barg J, McHale RJ, Ho MT, Ignatova E et al (1994). Transient down-regulation of neonatal rat brain mu-opioid receptors upon in utero exposure to buprenorphine. Brain Res Dev Brain Res 80: 158–162.
Bennett DS, Mohamed FB, Carmody DP, Bendersky M, Patel S, Khorrami M et al (2009). Response inhibition among early adolescents prenatally exposed to tobacco: an fMRI study. Neurotoxicol Teratol 31: 283–290.
Bennett DS, Mohamed FB, Carmody DP, Malik M, Faro SH, Lewis M (2013). Prenatal tobacco exposure predicts differential brain function during working memory in early adolescence: a preliminary investigation. Brain Imaging Behav 7: 49–59.
Betancourt LM, Yang W, Brodsky NL, Gallagher PR, Malmud EK, Giannetta JM et al (2011). Adolescents with and without gestational cocaine exposure: longitudinal analysis of inhibitory control, memory and receptive language. Neurotoxicol Teratol 33: 36–46.
Bhide PG (2009). Dopamine, cocaine and the development of cerebral cortical cytoarchitecture: a review of current concepts. Semin Cell Dev Biol 20: 395–402.
Bigsby R, LaGasse LL, Lester B, Shankaran S, Bada H, Bauer C et al (2011). Prenatal cocaine exposure and motor performance at 4 months. Am J Occup Ther 65: e60–e68.
Binder T, Vavrinkova B (2008). Prospective randomised comparative study of the effect of buprenorphine, methadone and heroin on the course of pregnancy, birthweight of newborns, early postpartum adaptation and course of the neonatal abstinence syndrome (NAS) in women followed up in the outpatient department. Neuro Endocrinol Lett 29: 80–86.
Bjorklund O, Kahlstrom J, Salmi P, Fredholm BB (2008). Perinatal caffeine, acting on maternal adenosine A(1) receptors, causes long-lasting behavioral changes in mouse offspring. PLoS One 3: e3977.
Bloch M, Althabe F, Onyamboko M, Kaseba-Sata C, Castilla EE, Freire S et al (2008). Tobacco use and secondhand smoke exposure during pregnancy: an investigative survey of women in 9 developing nations. Am J Public Health 98: 1833–1840.
Bodineau L, Cayetanot F, Sadani-Makki F, Bach V, Gros F, Lebleu A et al (2003). Consequences of in utero caffeine exposure on respiratory output in normoxic and hypoxic conditions and related changes of Fos expression: a study on brainstem-spinal cord preparations isolated from newborn rats. Pediatr Res 53: 266–273.
Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC et al (2011). A transient placental source of serotonin for the fetal forebrain. Nature 472: 347–350.
Bonnin A, Levitt P (2011). Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 197: 1–7 A landmark study demonstrating a new, direct role for 5-HT in maternal-placental-fetal interactions that could underlie the developmental impacts of 5-HT on long-lasting mental health outcomes.
Borgen LA, Davis WM, Pace HB (1971). Effects of synthetic 9 -tetrahydrocannabinol on pregnancy and offspring in the rat. Toxicol Appl Pharmacol 20: 480–486.
Bracken MB, Triche EW, Belanger K, Hellenbrand K, Leaderer BP (2003). Association of maternal caffeine consumption with decrements in fetal growth. Am J Epidemiol 157: 456–466.
Brent RL, Christian MS, Diener RM (2011). Evaluation of the reproductive and developmental risks of caffeine. Birth Defects Res B Dev Reprod Toxicol 92: 152–187.
Broening HW, Morford LL, Inman-Wood SL, Fukumura M, Vorhees CV (2001). 3,4-methylenedioxymethamphetamine (ecstasy)-induced learning and memory impairments depend on the age of exposure during early development. J Neurosci 21: 3228–3235.
Brown HL, Britton KA, Mahaffey D, Brizendine E, Hiett AK, Turnquest MA (1998). Methadone maintenance in pregnancy: a reappraisal. Am J Obstet Gynecol 179: 459–463.
Bruin JE, Gerstein HC, Holloway AC (2010). Long-term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci 116: 364–374 Discusses the issues involved with nicotine replacement therapy in pregnant women.
Brunell SC, Spear LP (2006). Effects of acute ethanol or amphetamine administration on the acoustic startle response and prepulse inhibition in adolescent and adult rats. Psychopharmacology (Berl) 186: 579–586.
Bubenikova-Valesova V, Kacer P, Syslova K, Rambousek L, Janovsky M, Schutova B et al (2009). Prenatal methamphetamine exposure affects the mesolimbic dopaminergic system and behavior in adult offspring. Int J Dev Neurosci 27: 525–530.
Bublitz MH, Stroud LR (2012). Maternal smoking during pregnancy and offspring brain structure and function: review and agenda for future research. Nicotine Tob Res 14: 388–397.
Buckley NE, Hansson S, Harta G, Mezey E (1998). Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 82: 1131–1149.
Budetti PP (1993). Achieving a uniform federal primary care policy. Opportunities presented by national health reform. JAMA 269: 498–501.
Bunikowski R, Grimmer I, Heiser A, Metze B, Schafer A, Obladen M (1998). Neurodevelopmental outcome after prenatal exposure to opiates. Eur J Pediatr 157: 724–730.
Buscariollo DL, Fang X, Greenwood V, Xue H, Rivkees SA, Wendler CC (2014). Embryonic caffeine exposure acts via a1 adenosine receptors to alter adult cardiac function and DNA methylation in mice. PLoS One 9: e87547.
Caldwell J, Dring LG, Williams RT (1972). Metabolism of ( 14 C)methamphetamine in man, the guinea pig and the rat. Biochem J 129: 11–22 Demonstrated how methamphetamine is metabolized in both animal and human models.
Campbell NG, Koprich JB, Kanaan NM, Lipton JW (2006). MDMA administration to pregnant Sprague-Dawley rats results in its passage to the fetal compartment. Neurotoxicol Teratol 28: 459–465 Demonstrated a strong correlation between fetal amniotic fluid and fetal brain—suggesting amniotic fluid could estimate fetal MDMA levels.
Campolongo P, Trezza V, Cassano T, Gaetani S, Morgese MG, Ubaldi M et al (2007). Perinatal exposure to delta-9-tetrahydrocannabinol causes enduring cognitive deficits associated with alteration of cortical gene expression and neurotransmission in rats. Addict Biol 12: 485–495.
Campolongo P, Trezza V, Palmery M, Trabace L, Cuomo V (2009). Developmental exposure to cannabinoids causes subtle and enduring neurofunctional alterations. Int Rev Neurobiol 85: 117–133 Excellent overview of the clinical and preclinical data on developmental marijuana exposure.
Canales JJ, Ferrer-Donato A (2014). Prenatal exposure to alcohol and 3,4-methylenedioxymethamphetamine (ecstasy) alters adult hippocampal neurogenesis and causes enduring memory deficits. Dev Neurosci 36: 10–17.
CARE Study Group (2008). Maternal caffeine intake during pregnancy and risk of fetal growth restriction: a large prospective observational study. BMJ 337: a2332. This large prospective longitudinal observational study found that caffeine consumption throughout pregnancy was associated with an increased risk of fetal growth restriction.
Carmody DP, Bennett DS, Lewis M (2011). The effects of prenatal cocaine exposure and gender on inhibitory control and attention. Neurotoxicol Teratol 33: 61–68 Study highlights the importance of considering gender specificity in prenatal cocaine effects.
Carter S, Paterson J, Gao W, Iusitini L (2008). Maternal smoking during pregnancy and behaviour problems in a birth cohort of 2-year-old Pacific children in New Zealand. Early Hum Dev 84: 59–66.
Chang L, Cloak C, Jiang CS, Farnham S, Tokeshi B, Buchthal S et al (2009). Altered neurometabolites and motor integration in children exposed to methamphetamine in utero. Neuroimage 48: 391–397.
Chang L, Smith LM, LoPresti C, Yonekura ML, Kuo J, Walot I et al (2004). Smaller subcortical volumes and cognitive deficits in children with prenatal methamphetamine exposure. Psychiatry Res 132: 95–106.
Chen R, Clifford A, Lang L, Anstey KJ (2013). Is exposure to secondhand smoke associated with cognitive parameters of children and adolescents?—a systematic literature review. Ann Epidemiol 23: 652–661.
Chiang YC, Hung TW, Ho IK (2013). Development of sensitization to methamphetamine in offspring prenatally exposed to morphine, methadone and buprenorphine. Addict Biol (e-pub ahead of print 3 April 2013). Outlined a potential mechanism for the effects of prenatal buprenorphine exposure on the effects of drugs of abuse.
Chiang YC, Hung TW, Lee CW, Yan JY, Ho IK (2010). Enhancement of tolerance development to morphine in rats prenatally exposed to morphine, methadone, and buprenorphine. J Biomed Sci 17: 46.
Chiriboga CA, Kuhn L, Wasserman GA (2007). Prenatal cocaine exposures and dose-related cocaine effects on infant tone and behavior. Neurotoxicol Teratol 29: 323–330.
Chistyakov V, Patkina N, Tammimaki A, Talka R, Salminen O, Belozertseva I et al (2010). Nicotine exposure throughout early development promotes nicotine self-administration in adolescent mice and induces long-lasting behavioural changes. Eur J Pharmacol 640: 87–93.
Cleary BJ, Eogan M, O'Connell MP, Fahey T, Gallagher PJ, Clarke T et al (2012). Methadone and perinatal outcomes: a prospective cohort study. Addiction 107: 1482–1492.
Cloak CC, Ernst T, Fujii L, Hedemark B, Chang L (2009). Lower diffusion in white matter of children with prenatal methamphetamine exposure. Neurology 72: 2068–2075.
Coleman T (2008). Reducing harm from tobacco smoke exposure during pregnancy. Birth Defects Res C Embryo Today 84: 73–79.
Coles CD, Platzman KA, Lynch ME, Freides D (2002). Auditory and visual sustained attention in adolescents prenatally exposed to alcohol. Alcohol Clin Exp Res 26: 263–271.
Conti AC, Young C, Olney JW, Muglia LJ (2009). Adenylyl cyclases types 1 and 8 promote pro-survival pathways after ethanol exposure in the neonatal brain. Neurobiol Dis 33: 111–118.
Cordeaux Y, Missfelder-Lobos H, Charnock-Jones DS, Smith GCS (2008). Stimulation of contractions in human myometrium by serotonin is unmasked by smooth muscle relaxants. Reprod Sci 15: 727–734.
Cornelius MD, Day NL (2009). Developmental consequences of prenatal tobacco exposure. Curr Opin Neurol 22: 121–125.
Cornelius MD, De Genna NM, Leech SL, Willford JA, Goldschmidt L, Day NL (2011). Effects of prenatal cigarette smoke exposure on neurobehavioral outcomes in 10-year-old children of adolescent mothers. Neurotoxicol Teratol 33: 137–144.
Cornelius MD, Goldschmidt L, DeGenna N, Day NL (2007). Smoking during teenage pregnancies: effects on behavioral problems in offspring. Nicotine Tob Res 9: 739–750.
Cornelius MD, Taylor PM, Geva D, Day NL (1995). Prenatal tobacco and marijuana use among adolescents: effects on offspring gestational age, growth, and morphology. Pediatrics 95: 738–743.
Covington CY, Nordstrom-Klee B, Ager J, Sokol R, Delaney-Black V (2002). Birth to age 7 growth of children prenatally exposed to drugs: a prospective cohort study. Neurotoxicol Teratol 24: 489–496.
Coyle MG, Salisbury AL, Lester BM, Jones HE, Lin H, Graf-Rohrmeister K et al (2012). Neonatal neurobehavior effects following buprenorphine versus methadone exposure. Addiction 107: 63–73.
Crandall JE, Hackett HE, Tobet SA, Kosofsky BE, Bhide PG (2004). Cocaine exposure decreases GABA neuron migration from the ganglionic eminence to the cerebral cortex in embryonic mice. Cereb Cortex 14: 665–675.
Czogala J, Goniewicz ML, Fidelus B, Zielinska-Danch W, Travers MJ, Sobczak A (2013). Secondhand exposure to vapors from electronic cigarettes. Nicotine Tob Res 16: 655–662.
da Silva RS, Hoffman A, de Souza DO, Lara DR, Bonan CD (2005). Maternal caffeine intake impairs MK-801-induced hyperlocomotion in young rats. Eur J Pharmacol 509: 155–159.
Dahl RE, Scher MS, Williamson DE, Robles N, Day N (1995). A longitudinal study of prenatal marijuana use. Effects on sleep and arousal at age 3 years. Arch Pediatr Adolesc Med 149: 145–150.
Day N, Sambamoorthi U, Taylor P, Richardson G, Robles N, Jhon Y et al (1991). Prenatal marijuana use and neonatal outcome. Neurotoxicol Teratol 13: 329–334.
Day NL, Goldschmidt L, Thomas CA (2006). Prenatal marijuana exposure contributes to the prediction of marijuana use at age 14. Addiction 101: 1313–1322.
Day NL, Richardson GA (1991). Prenatal alcohol exposure: a continuum of effects. Semin Perinatol 15: 271–279.
Day NL, Richardson GA (2004). An analysis of the effects of prenatal alcohol exposure on growth: a teratologic model. Am J Med Genet C Semin Med Genet 127C: 28–34 A seminal review on the effects of prenatal alcohol on growth demonstrating an interaction between the environment and prenatal exposure.
Day NL, Richardson GA, Goldschmidt L, Cornelius MD (2000). Effects of prenatal tobacco exposure on preschoolers' behavior. J Dev Behav Pediatr 21: 180–188.
Day NL, Richardson GA, Goldschmidt L, Robles N, Taylor PM, Stoffer DS et al (1994). Effect of prenatal marijuana exposure on the cognitive development of offspring at age three. Neurotoxicol Teratol 16: 169–175.
de Cubas MM, Field T (1993). Children of methadone-dependent women: developmental outcomes. Am J Orthopsychiatry 63: 266–276.
De Giorgio A, Comparini SE, Intra FS, Granato A (2012). Long-term alterations of striatal parvalbumin interneurons in a rat model of early exposure to alcohol. J Neurodev Disord 4: 18.
De Guio F, Mangin JF, Riviere D, Perrot M, Molteno CD, Jacobson SW et al (2014). A study of cortical morphology in children with fetal alcohol spectrum disorders. Hum Brain Mapp 35: 2285–2296.
de la Monte SM, Wands JR (2010). Role of central nervous system insulin resistance in fetal alcohol spectrum disorders. J Popul Ther Clin Pharmacol 17: e390–e404.
de la Torre R, Farre M, Roset PN, Pizarro N, Abanades S, Segura M et al (2004). Human pharmacology of MDMA: pharmacokinetics, metabolism, and disposition. Ther Drug Monit 26: 137–144.
De Montis GM, Devoto P, Angioi RM, Curreli V, Tagliamonte A (1983). In utero exposure to methadone produces a stable decrease of the cortex 5-HT transport system in rats. Eur J Pharmacol 90: 57–63.
De Vries TJ, Van Vliet BJ, Hogenboom F, Wardeh G, Van der Laan JW, Mulder AH et al (1991). Effect of chronic prenatal morphine treatment of mu-opioid receptor-regulated adenylate cyclase activity and neurotransmitter release in rat brain slices. Eur J Pharmacol 208: 97–104.
Dearlove JC, Betteridge TJ (1992). Stillbirth due to intravenous amphetamine. Br Med J 304: 548–548.
Della Grotta S, LaGasse LL, Arria AM, Derauf C, Grant P, Smith LM et al (2010). Patterns of methamphetamine use during pregnancy: results from the Infant Development, Environment, and Lifestyle (IDEAL) Study. Matern Child Health J 14: 519–527.
Derauf C, Lagasse LL, Smith LM, Newman E, Shah R, Neal CR et al (2012). Prenatal methamphetamine exposure and inhibitory control among young school-age children. J Pediatr 161: 452–459.
Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastiao AM (2013). Adenosine: setting the stage for plasticity. Trends Neurosci 36: 248–257.
DiNieri JA, Wang X, Szutorisz H, Spano SM, Kaur J, Casaccia P et al (2011). Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol Psychiatry 70: 763–769 Excellent example of clinical data with simultaneous support from an animal model.
Dixon SD, Bejar R (1989). Echoencephalographic findings in neonates associated with maternal cocaine and methamphetamine use—incidence and clinical correlates. J Pediatr 115: 770–778 Findings have revealed congenital anomalies in methamphetamine exposed neonates include cardiac anomalies, cranial abnormalities, and abnormal brain development closely resembling those in ill, asphyxiated infants.
Doremus TL, Brunell SC, Varlinskaya EI, Spear LP (2003). Anxiogenic effects during withdrawal from acute ethanol in adolescent and adult rats. Pharmacol Biochem Behav 75: 411–418.
Dow-Edwards D (2011). Translational issues for prenatal cocaine studies and the role of environment. Neurotoxicol Teratol 33: 9–16.
Dow-Edwards D, Iijima M, Stephenson S, Jackson A, Weedon J (2014). The effects of prenatal cocaine, post-weaning housing and sex on conditioned place preference in adolescent rats. Psychopharmacology (Berl) 231: 1543–1555.
Dwyer JB, Broide RS, Leslie FM (2008). Nicotine and brain development. Birth Defects Res C Embryo Today 84: 30–44.
Dwyer JB, McQuown SC, Leslie FM (2009). The dynamic effects of nicotine on the developing brain. Pharmacol Ther 122: 125–139.
Eckstrand KL, Ding Z, Dodge NC, Cowan RL, Jacobson JL, Jacobson SW et al (2012). Persistent dose-dependent changes in brain structure in young adults with low-to-moderate alcohol exposure in utero. Alcohol Clin Exp Res 36: 1892–1902 A detailed and comprehensive study following low-to-moderate alcohol exposure demonstrating significant and long-lasting changes in brain structure.
El Marroun H, Tiemeier H, Steegers EA, Jaddoe VW, Hofman A, Verhulst FC et al (2009). Intrauterine cannabis exposure affects fetal growth trajectories: the Generation R Study. J Am Acad Child Adolesc Psychiatry 48: 1173–1181.
England LJ, Levine RJ, Mills JL, Klebanoff MA, Yu KF, Cnattingius S (2003). Adverse pregnancy outcomes in snuff users. Am J Obstet Gynecol 189: 939–943.
Enters EK, Guo HZ, Pandey U, Ko DJ, Robinson SE (1991). The effect of prenatal methadone exposure on development and nociception during the early postnatal period of the rat. Neurotoxicol Teratol 13: 161–166.
Eriksson M, Jonsson B, Steneroth G, Zetterstrom R (1994). Cross-sectional growth of children whose mothers abused amphetamines during pregnancy. Acta Paediatr 83: 612–617.
Eriksson M, Jonsson B, Zetterstrom R (2000). Children of mothers abusing amphetamine: head circumference during infancy and psychosocial development until 14 years of age. Acta Paediatr 89: 1474–1478 Extensive follow-up data on affective, behavioral, and cognitive outcomes following prenatal amphetamines-exposed children from birth to age 14.
Eriksson PS, Ronnback L (1989). Effects of prenatal morphine treatment of rats on mortality, bodyweight and analgesic response in the offspring. Drug Alcohol Depend 24: 187–194.
Esposito ER, Horn KH, Greene RM, Pisano MM (2008). An animal model of cigarette smoke-induced in utero growth retardation. Toxicology 246: 193–202.
Espy KA, Fang H, Johnson C, Stopp C, Wiebe SA (2011). Prenatal tobacco exposure: developmental outcomes in the neonatal period. Dev Psychol 47: 153–156.
Fajemirokun-Odudeyi O, Sinha C, Tutty S, Pairaudeau P, Armstrong D, Phillips T et al (2006). Pregnancy outcome in women who use opiates. Eur J Obstet Gynecol Reprod Biol 126: 170–175.
Farber NB, Creeley CE, Olney JW (2010). Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol Dis 40: 200–206.
Farid WO, Dunlop SA, Tait RJ, Hulse GK (2008). The effects of maternally administered methadone, buprenorphine and naltrexone on offspring: review of human and animal data. Curr Neuropharmacol 6: 125–150.
Farid WO, Lawrence AJ, Krstew EV, Tait RJ, Hulse GK, Dunlop SA (2012). Maternally administered sustained-release naltrexone in rats affects offspring neurochemistry and behaviour in adulthood. PLoS One 7: e52812.
Fayyad J, De Graaf R, Kessler R, Alonso J, Angermeyer M, Demyttenaere K et al (2007). Cross-national prevalence and correlates of adult attention-deficit hyperactivity disorder. Br J Psychiatry 190: 402–409.
Ferguson v. City of Charleston (2001). Ferguson v. City of Charleston, No. 99-936, SUPREME COURT OF THE UNITED STATES.
Fergusson DM, Horwood LJ, Northstone K (2002). Maternal use of cannabis and pregnancy outcome. BJOG 109: 21–27.
Finger B, Schuetze P, Eiden RD (2014). Behavior problems among cocaine exposed children: role of physiological regulation and parenting. Neurotoxicol Teratol 42: 51–59.
Finnegan LP, Connaughton JF Jr, Kron RE, Emich JP (1975). Neonatal abstinence syndrome: assessment and management. Addict Dis 2: 141–158.
Fischer G, Johnson RE, Eder H, Jagsch R, Peternell A, Weninger M et al (2000). Treatment of opioid-dependent pregnant women with buprenorphine. Addiction 95: 239–244.
Flores G, de Jesus Gomez-Villalobos M, Rodriguez-Sosa L (2011). Prenatal amphetamine exposure effects on dopaminergic receptors and transporter in postnatal rats. Neurochem Res 36: 1740–1749.
Foltran F, Gregori D, Franchin L, Verduci E, Giovannini M (2011). Effect of alcohol consumption in prenatal life, childhood, and adolescence on child development. Nutr Rev 69: 642–659.
Ford DH, Rhines RK (1979). Prenatal exposure to methadone HCL in relationship to body and brain growth in the rat. Acta Neurol Scand 59: 248–262.
Frederick AL, Stanwood GD (2009). Drugs, biogenic amine targets and the developing brain. Dev Neurosci 31: 7–22.
Fried PA (1982). Marihuana use by pregnant women and effects on offspring: an update. Neurobehav Toxicol Teratol 4: 451–454.
Fried PA, Makin JE (1987). Neonatal behavioural correlates of prenatal exposure to marihuana, cigarettes and alcohol in a low risk population. Neurotoxicol Teratol 9: 1–7.
Fried PA, O'Connell CM, Watkinson B (1992a). 60- and 72-month follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol: cognitive and language assessment. J Dev Behav Pediatr 13: 383–391.
Fried PA, Smith AM (2001). A literature review of the consequences of prenatal marihuana exposure. An emerging theme of a deficiency in aspects of executive function. Neurotoxicol Teratol 23: 1–11 Comprehensive review of how prenatal marijuana affects cognition and executive function.
Fried PA, Watkinson B (1988). 12- and 24-month neurobehavioural follow-up of children prenatally exposed to marihuana, cigarettes and alcohol. Neurotoxicol Teratol 10: 305–313.
Fried PA, Watkinson B (1990). 36- and 48-month neurobehavioral follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol. J Dev Behav Pediatr 11: 49–58.
Fried PA, Watkinson B (2000). Visuoperceptual functioning differs in 9- to 12-year olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol 22: 11–20.
Fried PA, Watkinson B, Dillon RF, Dulberg CS (1987). Neonatal neurological status in a low-risk population after prenatal exposure to cigarettes, marijuana, and alcohol. J Dev Behav Pediatr 8: 318–326.
Fried PA, Watkinson B, Gray R (1992b). A follow-up study of attentional behavior in 6-year-old children exposed prenatally to marihuana, cigarettes, and alcohol. Neurotoxicol Teratol 14: 299–311.
Fried PA, Watkinson B, Gray R (1998). Differential effects on cognitive functioning in 9- to 12-year olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol 20: 293–306.
Fried PA, Watkinson B, Gray R (1999). Growth from birth to early adolescence in offspring prenatally exposed to cigarettes and marijuana. Neurotoxicol Teratol 21: 513–525.
Fried PA, Watkinson B, Gray R (2003). Differential effects on cognitive functioning in 13- to 16-year-olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol 25: 427–436.
Fried PA, Watkinson B, Willan A (1984). Marijuana use during pregnancy and decreased length of gestation. Am J Obstet Gynecol 150: 23–27.
Gabriel M, Taylor C, Burhans L (2003). In utero cocaine, discriminative avoidance learning with low-salient stimuli and learning-related neuronal activity in rabbits (Oryctolagus cuniculus). Behav Neurosci 117: 912–926.
Galineau L, Belzung C, Kodas E, Bodard S, Guilloteau D, Chalon S (2005). Prenatal 3,4-methylenedioxymethamphetamine (ecstasy) exposure induces long-term alterations in the dopaminergic and serotonergic functions in the rat. Brain Res Dev Brain Res 154: 165–176.
Garavan H, Morgan RE, Mactutus CF, Levitsky DA, Booze RM, Strupp BJ (2000). Prenatal cocaine exposure impairs selective attention: evidence from serial reversal and extradimensional shift tasks. Behav Neurosci 114: 725–738.
Gatzke-Kopp LM, Beauchaine TP (2007). Direct and passive prenatal nicotine exposure and the development of externalizing psychopathology. Child Psychiatry Hum Dev 38: 255–269.
Gill AC, Oei J, Lewis NL, Younan N, Kennedy I, Lui K (2003). Strabismus in infants of opiate-dependent mothers. Acta Paediatr 92: 379–385.
Goldschmidt L, Day NL, Richardson GA (2000). Effects of prenatal marijuana exposure on child behavior problems at age 10. Neurotoxicol Teratol 22: 325–336.
Goldschmidt L, Richardson GA, Willford J, Day NL (2008). Prenatal marijuana exposure and intelligence test performance at age 6. J Am Acad Child Adolesc Psychiatry 47: 254–263.
Goldschmidt L, Richardson GA, Willford JA, Severtson SG, Day NL (2012). School achievement in 14-year-old youths prenatally exposed to marijuana. Neurotoxicol Teratol 34: 161–167.
Goodlett CR, Horn KH, Zhou FC (2005). Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (Maywood) 230: 394–406.
Gouin K, Murphy K, Shah PS (2011). Effects of cocaine use during pregnancy on low birthweight and preterm birth: systematic review and metaanalyses. Am J Obstet Gynecol 204: 340, e341-312.
Graf WD, Kekatpure MV, Kosofsky BE (2013). Prenatal-onset neurodevelopmental disorders secondary to toxins, nutritional deficiencies, and maternal illness. Handb Clin Neurol 111: 143–159.
Granato A (2006). Altered organization of cortical interneurons in rats exposed to ethanol during neonatal life. Brain Res 1069: 23–30.
Granato A, Di Rocco F, Zumbo A, Toesca A, Giannetti S (2003). Organization of cortico-cortical associative projections in rats exposed to ethanol during early postnatal life. Brain Res Bull 60: 339–344.
Granato A, Palmer LM, De Giorgio A, Tavian D, Larkum ME (2012). Early exposure to alcohol leads to permanent impairment of dendritic excitability in neocortical pyramidal neurons. J Neurosci 32: 1377–1382 A detailed, mechanistic analysis of the effects of early postnatal ethanol in the rat (similar to third trimester in human) on cortical neuron excitability. Dual whole-cell recordings from the soma and distal apical dendrites revealed a lower number and a shorter duration of dendritic spikes ethanol-treated animals, resulting in reduced action potentials recorded at the soma after dendritic current injection.
Granato A, Van Pelt J (2003). Effects of early ethanol exposure on dendrite growth of cortical pyramidal neurons: inferences from a computational model. Brain Res Dev Brain Res 142: 223–227.
Gray KA, Day NL, Leech S, Richardson GA (2005). Prenatal marijuana exposure: effect on child depressive symptoms at ten years of age. Neurotoxicol Teratol 27: 439–448.
Gressens P, Mesples B, Sahir N, Marret S, Sola A (2001). Environmental factors and disturbances of brain development. Semin Neonatol 6: 185–194.
Griffith DR, Azuma SD, Chasnoff IJ (1994). Three-year outcome of children exposed prenatally to drugs. J Am Acad Child Adolesc Psychiatry 33: 20–27.
Guo HZ, Enters EK, McDowell KP, Robinson SE (1990). The effect of prenatal exposure to methadone on neurotransmitters in neonatal rats. Brain Res Dev Brain Res 57: 296–298.
Guo X, Spencer JW, Suess PE, Hickey JE, Better WE, Herning RI (1994). Cognitive brain potential alterations in boys exposed to opiates: in utero and lifestyle comparisons. Addict Behav 19: 429–441.
Gupta M, Mulvihill AO, Lascaratos G, Fleck BW, George ND (2012). Nystagmus and reduced visual acuity secondary to drug exposure in utero: long-term follow-up. J Pediatr Ophthalmol Strabismus 49: 58–63.
Hamilton LR, Czoty PW, Gage HD, Nader MA (2010). Characterization of the dopamine receptor system in adult rhesus monkeys exposed to cocaine throughout gestation. Psychopharmacology (Berl) 210: 481–488.
Hammer RP Jr, Seatriz JV, Ricalde AR (1991). Regional dependence of morphine-induced mu-opiate receptor down-regulation in perinatal rat brain. Eur J Pharmacol 209: 253–256.
Hans SL (1989). Developmental consequences of prenatal exposure to methadone. Ann N Y Acad Sci 562: 195–207 Suggested a major role often overlooked in previously published studies of socioeconomic status of control and exposed groups greatly confounding outcomes.
Harlan RE, Song DD (1994). Prenatal morphine treatment and the development of the striatum. Regul Pept 54: 117–118.
Harrod SB, Lacy RT, Morgan AJ (2012). Offspring of prenatal IV nicotine exposure exhibit increased sensitivity to the reinforcing effects of methamphetamine. Front Pharmacol 3: 116.
Harvey JA (2004). Cocaine effects on the developing brain: current status. Neurosci Biobehav Rev 27: 751–764.
Harvey JA, Romano AG, Gabriel M, Simansky KJ, Du W, Aloyo VJ et al (2001). Effects of prenatal exposure to cocaine on the developing brain: anatomical, chemical, physiological and behavioral consequences. Neurotox Res 3: 117–143.
Hashimoto-Torii K, Kawasawa YI, Kuhn A, Rakic P (2011). Combined transcriptome analysis of fetal human and mouse cerebral cortex exposed to alcohol. Proc Natl Acad Sci USA 108: 4212–4217.
Hauser KF, McLaughlin PJ, Zagon IS (1987). Endogenous opioids regulate dendritic growth and spine formation in developing rat brain. Brain Res 416: 157–161.
Hauser KF, McLaughlin PJ, Zagon IS (1989). Endogenous opioid systems and the regulation of dendritic growth and spine formation. J Comp Neurol 281: 13–22.
Heath CJ, King SL, Gotti C, Marks MJ, Picciotto MR (2010). Cortico-thalamic connectivity is vulnerable to nicotine exposure during early postnatal development through alpha4/beta2/alpha5 nicotinic acetylcholine receptors. Neuropsychopharmacology 35: 2324–2338 Identifies not only a particular behavior associated with prenatal nicotine exposure, but also the mechanism, circuitry, and critical exposure period.
Hellemans KG, Verma P, Yoon E, Yu WK, Young AH, Weinberg J (2010). Prenatal alcohol exposure and chronic mild stress differentially alter depressive- and anxiety-like behaviors in male and female offspring. Alcohol Clin Exp Res 34: 633–645.
Hickey JE, Suess PE, Newlin DB, Spurgeon L, Porges SW (1995). Vagal tone regulation during sustained attention in boys exposed to opiates in utero. Addict Behav 20: 43–59.
Hicks SD, Middleton FA, Miller MW (2010). Ethanol-induced methylation of cell cycle genes in neural stem cells. J Neurochem 114: 1767–1780.
Himes SK, Stroud LR, Scheidweiler KB, Niaura RS, Huestis MA (2013). Prenatal tobacco exposure, biomarkers for tobacco in meconium, and neonatal growth outcomes. J Pediatr 162: 970–975.
Ho E, Karimi-Tabesh L, Koren G (2001). Characteristics of pregnant women who use ecstasy (3, 4-methylenedioxymethamphetamine). Neurotoxicol Teratol 23: 561–567.
Hohmann CF (2003). A morphogenetic role for acetylcholine in mouse cerebral neocortex. Neurosci Biobehav Rev 27: 351–363.
Hou Y, Tan Y, Belcheva MM, Clark AL, Zahm DS, Coscia CJ (2004). Differential effects of gestational buprenorphine, naloxone, and methadone on mesolimbic mu opioid and ORL1 receptor G protein coupling. Brain Res Dev Brain Res 151: 149–157.
Hovious JR, Peters MA (1984). Analgesic effect of opiates in offspring of opiate-treated female rats. Pharmacol Biochem Behav 21: 555–559.
Howell KK, Lynch ME, Platzman KA, Smith GH, Coles CD (2006). Prenatal alcohol exposure and ability, academic achievement, and school functioning in adolescence: a longitudinal follow-up. J Pediatr Psychol 31: 116–126.
Hoyme HE, May PA, Kalberg WO, Kodituwakku P, Gossage JP, Trujillo PM et al (2005). A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: clarification of the 1996 institute of medicine criteria. Pediatrics 115: 39–47.
Huang LZ, Liu X, Griffith WH, Winzer-Serhan UH (2007). Chronic neonatal nicotine increases anxiety but does not impair cognition in adult rats. Behav Neurosci 121: 1342–1352.
Hulse G, O’Neil G (2002). Using naltrexone implants in the management of the pregnant heroin user. Aust N Z J Obstet Gynaecol 42: 569–573.
Hulse GK, Arnold-Reed DE, O’Neil G, Hansson RC (2003). Naltrexone implant and blood naltrexone levels over pregnancy. Aust N Z J Obstet Gynaecol 43: 386–388.
Hulse GK, Milne E, English DR, Holman CD (1997). The relationship between maternal use of heroin and methadone and infant birth weight. Addiction 92: 1571–1579.
Hulse GK, Milne E, English DR, Holman CD (1998). Assessing the relationship between maternal opiate use and neonatal mortality. Addiction 93: 1033–1042.
Hulse GK, O’Neil G, Arnold-Reed DE (2004). Methadone maintenance vs. implantable naltrexone treatment in the pregnant heroin user. Int J Gynaecol Obstet 85: 170–171.
Hulse GK, O’Neill G, Pereira C, Brewer C (2001). Obstetric and neonatal outcomes associated with maternal naltrexone exposure. Aust N Z J Obstet Gynaecol 41: 424–428 One of the first clinical reports of prenatal naltrexone exposure.
Hunt RW, Tzioumi D, Collins E, Jeffery HE (2008). Adverse neurodevelopmental outcome of infants exposed to opiate in-utero. Early Hum Dev 84: 29–35.
Hurd YL, Wang X, Anderson V, Beck O, Minkoff H, Dow-Edwards D (2005). Marijuana impairs growth in mid-gestation fetuses. Neurotoxicol Teratol 27: 221–229.
Hurt RD, Renner CC, Patten CA, Ebbert JO, Offord KP, Schroeder DR et al (2005). Iqmik—a form of smokeless tobacco used by pregnant Alaska natives: nicotine exposure in their neonates. J Matern Fetal Neonatal Med 17: 281–289.
Hutchings DE, Hamowy AS, Williams EM, Zmitrovich AC (1996). Prenatal administration of buprenorphine in the rat: effects on the rest-activity cycle at 22 and 30 days of age. Pharmacol Biochem Behav 55: 607–613.
Hutchings DE, Martin BR, Gamagaris Z, Miller N, Fico T (1989). Plasma concentrations of delta-9-tetrahydrocannabinol in dams and fetuses following acute or multiple prenatal dosing in rats. Life Sci 44: 697–701.
Hutchings DE, Zmitrovich A, Brake SC, Malowany D, Church S, Nero TJ (1992). Prenatal administration of methadone using the osmotic minipump: effects on maternal and offspring toxicity, growth, and behavior in the rat. Neurotoxicol Teratol 14: 65–71.
Hutchings DE, Zmitrovich AC, Brake SC, Church SH, Malowany D (1993). Prenatal administration of methadone in the rat increases offspring acoustic startle amplitude at age 3 weeks. Neurotoxicol Teratol 15: 157–164.
Indredavik MS, Brubakk AM, Romundstad P, Vik T (2007). Prenatal smoking exposure and psychiatric symptoms in adolescence. Acta Paediatr 96: 377–382.
Institute of Medicine (1996) Fetal Alcohol Syndrome:Diagnosis, Epidemiology, Prevention, and Treatment. The National Academies Press: Washington DC.
Jacobsen LK, Slotkin TA, Mencl WE, Frost SJ, Pugh KR (2007). Gender-specific effects of prenatal and adolescent exposure to tobacco smoke on auditory and visual attention. Neuropsychopharmacology 32: 2453–2464.
Jacobsen LK, Slotkin TA, Westerveld M, Mencl WE, Pugh KR (2006). Visuospatial memory deficits emerging during nicotine withdrawal in adolescents with prenatal exposure to active maternal smoking. Neuropsychopharmacology 31: 1550–1561.
Jacobson JL, Jacobson SW (1999). Drinking moderately and pregnancy. Effects on child development. Alcohol Res Health 23: 25–30.
Jacobson JL, Jacobson SW, Sokol RJ, Martier SS, Ager JW, Kaplan-Estrin MG (1993). Teratogenic effects of alcohol on infant development. Alcohol Clin Exp Res 17: 174–183.
Jansson LM, Velez M, Harrow C (2009). The opioid-exposed newborn: assessment and pharmacologic management. J Opioid Manag 5: 47–55.
Jeng W, Wong AW, Ting AKR, Wells PG (2005). Methamphetamine-enhanced embryonic oxidative DNA damage and neurodevelopmental deficits. Free Radic Biol Med 39: 317–326.
Jones HE, Chisolm MS, Jansson LM, Terplan M (2013). Naltrexone in the treatment of opioid-dependent pregnant women: the case for a considered and measured approach to research. Addiction 108: 233–247.
Jones J, Rios R, Jones M, Lewis D, Plate C (2009). Determination of amphetamine and methamphetamine in umbilical cord using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877: 3701–3706.
Jones KL (1986). Fetal alcohol syndrome. Pediatr Rev 8: 122–126.
Jones LB, Stanwood GD, Reinoso BS, Washington RA, Wang HY, Friedman E et al (2000). In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J Neurosci 20: 4606–4614.
Joya X, Pujadas M, Falcon M, Civit E, Garcia-Algar O, Vall O et al (2010). Gas chromatography-mass spectrometry assay for the simultaneous quantification of drugs of abuse in human placenta at 12th week of gestation. Forensic Sci Int 196: 38–42 Used a chromatography-mass spectrometry assay for the simultaneous quantification of drugs of abuse—including amphetamine, methamphetamine, MDMA, methadone, cocaine, morphine, nicotine, and cotinine—in human placenta at 12th week of gestation.
Jutras-Aswad D, DiNieri JA, Harkany T, Hurd YL (2009). Neurobiological consequences of maternal cannabis on human fetal development and its neuropsychiatric outcome. Eur Arch Psychiatry Clin Neurosci 259: 395–412.
Kabir ZD, McCarthy DM, Bhide PG, Kosofsky BE (2013). Cup of Joe: a brain development ‘no’? Sci Transl Med 5: 197fs130.
Kable JA, Coles CD, Lynch ME, Carroll J (2009). The impact of maternal smoking on fast auditory brainstem responses. Neurotoxicol Teratol 31: 216–224.
Kaltenbach K, Berghella V, Finnegan L (1998). Opioid dependence during pregnancy. Effects and management. Obstet Gynecol Clin North Am 25: 139–151.
Kandall SR, Albin S, Lowinson J, Berle B, Eidelman AI, Gartner LM (1976). Differential effects of maternal heroin and methadone use on birthweight. Pediatrics 58: 681–685.
Katayama H, Itami S, Koizumi H, Tsutsui M (1987). Epidermal cell culture using Sephadex beads coated with denatured collagen (cytodex 3). J Invest Dermatol 88: 33–36.
Kater SB, Lipton SA (1995). Neurotransmitter regulation of neuronal outgrowth, plasticity and survival in the year 2001. Trends Neurosci 18: 71–72.
Keating E, Goncalves P, Campos I, Costa F, Martel F (2009). Folic acid uptake by the human syncytiotrophoblast: interference by pharmacotherapy, drugs of abuse and pathological conditions. Reprod Toxicol 28: 511–520.
Keimpema E, Mackie K, Harkany T (2011). Molecular model of cannabis sensitivity in developing neuronal circuits. Trends Pharmacol Sci 32: 551–561.
Key AP, Ferguson M, Molfese DL, Peach K, Lehman C, Molfese VJ (2007). Smoking during pregnancy affects speech-processing ability in newborn infants. Environ Health Perspect 115: 623–629.
Killinger CE, Robinson S, Stanwood GD (2012). Subtle biobehavioral effects produced by paternal cocaine exposure. Synapse 66: 902–908.
Kirby ML (1983). Changes in [3H]naloxone binding in spinal cord of rats treated prenatally with morphine. Neuropharmacology 22: 303–307.
Kirby ML, Holtzmann SG (1982). Effects of chronic opiate administration of spontaneous activity of fetal rats. Pharmacol Biochem Behav 16: 263–269.
Kollins SH, McClernon FJ, Fuemmeler BF (2005). Association between smoking and attention-deficit/hyperactivity disorder symptoms in a population-based sample of young adults. Arch Gen Psychiatry 62: 1142–1147.
Koprich JB, Campbell NG, Lipton JW (2003a). Neonatal 3,4-methylenedioxymethamphetamine (ecstasy) alters dopamine and serotonin neurochemistry and increases brain-derived neurotrophic factor in the forebrain and brainstem of the rat. Brain Res Dev Brain Res 147: 177–182.
Koprich JB, Chen EY, Kanaan NM, Campbell NG, Kordower JH, Lipton JW (2003b). Prenatal 3,4-methylenedioxymethamphetamine (ecstasy) alters exploratory behavior, reduces monoamine metabolism, and increases forebrain tyrosine hydroxylase fiber density of juvenile rats. Neurotoxicol Teratol 25: 509–517.
Kotimaa AJ, Moilanen I, Taanila A, Ebeling H, Smalley SL, McGough JJ et al (2003). Maternal smoking and hyperactivity in 8-year-old children. J Am Acad Child Adolesc Psychiatry 42: 826–833.
Kuwahara MD, Sparber SB (1981). Prenatal withdrawal from opiates interferes with hatching of otherwise viable chick fetuses. Science 212: 945–947.
Ladhani NN, Shah PS, Murphy KE (2011). Prenatal amphetamine exposure and birth outcomes: a systematic review and metaanalysis. Am J Obstet Gynecol 205: 219 e211-217. An extensive meta-analysis of 10 studies of low-to-moderate risk of bias submits that AMPH exposure in pregnancy is associated with higher odds of preterm birth, low birth weight, and small size for gestational age.
LaGasse LL, Derauf C, Smith LM, Newman E, Shah R, Neal C et al (2012). Prenatal methamphetamine exposure and childhood behavior problems at 3 and 5 years of age. Pediatrics 129: 681–688.
Lambers DS, Clark KE (1996). The maternal and fetal physiologic effects of nicotine. Semin Perinatol 20: 115–126.
Langley K, Heron J, Smith GD, Thapar A (2012). Maternal and paternal smoking during pregnancy and risk of ADHD symptoms in offspring: testing for intrauterine effects. Am J Epidemiol 176: 261–268.
Langley K, Holmans PA, van den Bree MB, Thapar A (2007). Effects of low birth weight, maternal smoking in pregnancy and social class on the phenotypic manifestation of Attention Deficit Hyperactivity Disorder and associated antisocial behaviour: investigation in a clinical sample. BMC Psychiatry 7: 26.
Lauder JM (1993). Neurotransmitters as growth regulatory signals: role of receptors and second messengers. Trends Neurosci 16: 233–240 A classic review by a pioneer in the field, describing how neurotransmitters can regulate morphogenetic activities such as proliferation, differentiation, cell motility and metamorphosis.
Lawrence J, Xiao D, Xue Q, Rejali M, Yang S, Zhang L (2008). Prenatal nicotine exposure increases heart susceptibility to ischemia/reperfusion injury in adult offspring. J Pharmacol Exp Ther 324: 331–341.
Lebel C, Warner T, Colby J, Soderberg L, Roussotte F, Behnke M et al (2013). White matter microstructure abnormalities and executive function in adolescents with prenatal cocaine exposure. Psychiatry Res 213: 161–168.
Leech SL, Richardson GA, Goldschmidt L, Day NL (1999). Prenatal substance exposure: effects on attention and impulsivity of 6-year-olds. Neurotoxicol Teratol 21: 109–118.
Lejeune C, Simmat-Durand L, Gourarier L, Aubisson S (2006). Prospective multicenter observational study of 260 infants born to 259 opiate-dependent mothers on methadone or high-dose buprenophine substitution. Drug Alcohol Depend 82: 250–257.
Lenoir D, Barg J, Simantov R (1984). Characterization and down-regulation of opiate receptors in aggregating fetal rat brain cells. Brain Res 304: 285–290.
Leon D, Albasanz JL, Ruiz MA, Martin M (2005). Chronic caffeine or theophylline intake during pregnancy inhibits A1 receptor function in the rat brain. Neuroscience 131: 481–489.
Lester BM, Dreher M (1989). Effects of marijuana use during pregnancy on newborn cry. Child Dev 60: 765–771.
Lester BM, Padbury JF (2009). Third pathophysiology of prenatal cocaine exposure. Dev Neurosci 31: 23–35 Effects of cocaine on fetal development may also act by altering genetic programming causing acute and long-term adverse effects.
Lester BM, Tronick EZ, LaGasse L, Seifer R, Bauer CR, Shankaran S et al (2002). The maternal lifestyle study: effects of substance exposure during pregnancy on neurodevelopmental outcome in 1-month-old infants. Pediatrics 110: 1182–1192.
Levin ED, Briggs SJ, Christopher NC, Rose JE (1993). Prenatal nicotine exposure and cognitive performance in rats. Neurotoxicol Teratol 15: 251–260.
Levin ED, Lawrence S, Petro A, Horton K, Seidler FJ, Slotkin TA (2006). Increased nicotine self-administration following prenatal exposure in female rats. Pharmacol Biochem Behav 85: 669–674.
Levine TP, Liu J, Das A, Lester B, Lagasse L, Shankaran S et al (2008). Effects of prenatal cocaine exposure on special education in school-aged children. Pediatrics 122: e83–e91.
Lewis BA, Kirchner HL, Short EJ, Minnes S, Weishampel P, Satayathum S et al (2007). Prenatal cocaine and tobacco effects on children's language trajectories. Pediatrics 120: e78–e85.
Lewis BA, Minnes S, Short EJ, Weishampel P, Satayathum S, Min MO et al (2011). The effects of prenatal cocaine on language development at 10 years of age. Neurotoxicol Teratol 33: 17–24.
Li Z, Santhanam P, Coles CD, Ellen Lynch M, Hamann S, Peltier S et al (2013). Prenatal cocaine exposure alters functional activation in the ventral prefrontal cortex and its structural connectivity with the amygdala. Psychiatry Res 213: 47–55.
Lichtblau L, Sparber SB (1981). Opiate withdrawal in utero increases neonatal morbidity in the rat. Science 212: 943–945.
Lidow MS, Koh PO, Arnsten AF (2003). D1 dopamine receptors in the mouse prefrontal cortex: immunocytochemical and cognitive neuropharmacological analyses. Synapse 47: 101–108.
Lindblad F, Hjern A (2010). ADHD after fetal exposure to maternal smoking. Nicotine Tob Res 12: 408–415.
Linn S, Schoenbaum SC, Monson RR, Rosner R, Stubblefield PC, Ryan KJ (1983). The association of marijuana use with outcome of pregnancy. Am J Public Health 73: 1161–1164.
Little BB, Snell LM, Gilstrap LC 3rd (1988). Methamphetamine abuse during pregnancy: outcome and fetal effects. Obstet Gynecol 72: 541–544.
Litto WJ, Griffin JP, Rabii J (1983). Influence of morphine during pregnancy on neuroendocrine regulation of pituitary hormone secretion. J Endocrinol 98: 289–295.
Liu J, Lester BM, Neyzi N, Sheinkopf SJ, Gracia L, Kekatpure M et al (2013). Regional brain morphometry and impulsivity in adolescents following prenatal exposure to cocaine and tobacco. JAMA Pediatr 167: 348–354.
Lloyd SA, Faherty CJ, Smeyne RJ (2006). Adult and in utero exposure to cocaine alters sensitivity to the Parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience 137: 905–913.
Long DT, King MA, Sheehan J (1992). Comparative evaluation of image segmentation methods for volume quantitation in SPECT. Med Phys 19: 483–489.
Loomans EM, Hofland L, van der Stelt O, van der Wal MF, Koot HM, Van den Bergh BR et al (2012). Caffeine intake during pregnancy and risk of problem behavior in 5- to 6-year-old children. Pediatrics 130: e305–e313.
Lorenzo AM, Leon D, Castillo CA, Ruiz MA, Albasanz JL, Martin M (2010). Maternal caffeine intake during gestation and lactation down-regulates adenosine A1 receptor in rat brain from mothers and neonates. J Neurosci Res 88: 1252–1261.
Lotfipour S, Ferguson E, Leonard G, Perron M, Pike B, Richer L et al (2009). Orbitofrontal cortex and drug use during adolescence: role of prenatal exposure to maternal smoking and BDNF genotype. Arch Gen Psychiatry 66: 1244–1252 Report indicating that prenatal cigarette exposure produces long-term behavioral and neurological aberrations.
Lotfipour S, Leonard G, Perron M, Pike B, Richer L, Seguin JR et al (2010). Prenatal exposure to maternal cigarette smoking interacts with a polymorphism in the alpha6 nicotinic acetylcholine receptor gene to influence drug use and striatum volume in adolescence. Mol Psychiatry 15: 6–8.
Lu R, Liu X, Long H, Ma L (2012). Effects of prenatal cocaine and heroin exposure on neuronal dendrite morphogenesis and spatial recognition memory in mice. Neurosci Lett 522: 128–133.
Luo J, Miller MW (1998). Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Res Brain Res Rev 27: 157–167.
Lupton C, Burd L, Harwood R (2004). Cost of fetal alcohol spectrum disorders. Am J Med Genet C Semin Med Genet 127C: 42–50.
Maas U, Kattner E, Weingart-Jesse B, Schafer A, Obladen M (1990). Infrequent neonatal opiate withdrawal following maternal methadone detoxification during pregnancy. J Perinat Med 18: 111–118.
Mactutus CF, Herman AS, Booze RM (1994). Chronic intravenous model for studies of drug (Ab)use in the pregnant and/or group-housed rat: an initial study with cocaine. Neurotoxicol Teratol 16: 183–191 The first rodent intravenous cocaine during pregnancy animal model.
Malanga CJ 3rd, Kosofsky BE (1999). Mechanisms of action of drugs of abuse on the developing fetal brain. Clin Perinatol 26: 17–37 v-vi.
Mansi G, Raimondi F, Pichini S, Capasso L, Sarno M, Zuccaro P et al (2007). Neonatal urinary cotinine correlates with behavioral alterations in newborns prenatally exposed to tobacco smoke. Pediatr Res 61: 257–261.
Mantha K, Laufer BI, Singh SM (2014). Molecular changes during neurodevelopment following second-trimester binge ethanol exposure in a mouse model of fetal alcohol spectrum disorder: from immediate effects to long-term adaptation. Dev Neurosci 36: 29–43.
Martinez A, Kastner B, Taeusch HW (1999). Hyperphagia in neonates withdrawing from methadone. Arch Dis Child Fetal Neonatal Ed 80: F178–F182.
Mattson SN, Riley EP, Gramling L, Delis DC, Jones KL (1998). Neuropsychological comparison of alcohol-exposed children with or without physical features of fetal alcohol syndrome. Neuropsychology 12: 146–153.
Mayes LC (1994). Neurobiology of prenatal cocaine exposure effect on developing monoamine systems. Infant Mental Hlth J 15: 121–133.
Mayes LC (2002). A behavioral teratogenic model of the impact of prenatal cocaine exposure on arousal regulatory systems. Neurotoxicol Teratol 24: 385–395.
Mayes LC, Cicchetti D, Acharyya S, Zhang H (2003). Developmental trajectories of cocaine-and-other-drug-exposed and non-cocaine-exposed children. J Dev Behav Pediatr 24: 323–335.
McCarthy DM, Bhide PG (2012). Prenatal cocaine exposure decreases parvalbumin-immunoreactive neurons and GABA-to-projection neuron ratio in the medial prefrontal cortex. Dev Neurosci 34: 174–183.
McCarthy DM, Brown AN, Bhide PG (2012). Regulation of BDNF expression by cocaine. Yale J Biol Med 85: 437–446.
McElhatton PR, Bateman DN, Evans C, Pughe KR, Thomas SH (1999). Congenital anomalies after prenatal ecstasy exposure. Lancet 354: 1441–1442.
McGinty JF, Ford DH (1980). Effects of prenatal methadone on rat brain catecholamines. Dev Neurosci 3: 224–234.
McKnight v. State of South Carolina (2008). McKnight v. State of South Carolina,. 26484, THE STATE OF SOUTH CAROLINA In The Supreme Court.
McLaughlin AA, Minnes S, Singer LT, Min M, Short EJ, Scott TL et al (2011). Caregiver and self-report of mental health symptoms in 9-year old children with prenatal cocaine exposure. Neurotoxicol Teratol 33: 582–591.
McLaughlin PJ, Tobias SW, Lang CM, Zagon IS (1997a). Chronic exposure to the opioid antagonist naltrexone during pregnancy: maternal and offspring effects. Physiol Behav 62: 501–508.
McLaughlin PJ, Tobias SW, Lang CM, Zagon IS (1997b). Opioid receptor blockade during prenatal life modifies postnatal behavioral development. Pharmacol Biochem Behav 58: 1075–1082.
McLaughlin PJ, Zagon IS, White WJ (1978). Perinatal methadone exposure in rats. Effects on body and organ development. Biol Neonate 34: 48–54.
Medina AE, Krahe TE, Ramoa AS (2006). Restoration of neuronal plasticity by a phosphodiesterase type 1 inhibitor in a model of fetal alcohol exposure. J Neurosci 26: 1057–1060.
Medina Jimenez M, Lujan Estrada M, Rodriguez R (1997). Influence of chronic prenatal and postnatal administration of naltrexone in locomotor activity induced by morphine in mice. Arch Med Res 28: 61–65.
Mehmedic Z, Chandra S, Slade D, Denham H, Foster S, Patel AS et al (2010). Potency trends of Delta9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 55: 1209–1217.
Mereu G, Fa M, Ferraro L, Cagiano R, Antonelli T, Tattoli M et al (2003). Prenatal exposure to a cannabinoid agonist produces memory deficits linked to dysfunction in hippocampal long-term potentiation and glutamate release. Proc Natl Acad Sci USA 100: 4915–4920.
Meyer JS, Grande M, Johnson K, Ali SF (2004). Neurotoxic effects of MDMA (‘ecstasy’) administration to neonatal rats. Int J Dev Neurosci 22: 261–271.
Meyer KD, Zhang H, Zhang L (2009). Prenatal cocaine exposure abolished ischemic preconditioning-induced protection in adult male rat hearts: role of PKCepsilon. Am J Physiol Heart Circ Physiol 296: H1566–H1576.
Meyer KD, Zhang L (2009). Short- and long-term adverse effects of cocaine abuse during pregnancy on the heart development. Ther Adv Cardiovasc Dis 3: 7–16.
Miller MW (2007). Exposure to ethanol during gastrulation alters somatosensory-motor cortices and the underlying white matter in the macaque. Cereb Cortex 17: 2961–2971.
Miller MW, Hu H (2009). Lability of neuronal lineage decisions is revealed by acute exposures to ethanol. Dev Neurosci 31: 50–57.
Minnes S, Robin NH, Alt AA, Kirchner HL, Satayathum S, Salbert BA et al (2006). Dysmorphic and anthropometric outcomes in 6-year-old prenatally cocaine-exposed children. Neurotoxicol Teratol 28: 28–38.
Minnes S, Singer LT, Min MO, Lang AM, Ben-Harush A, Short E et al (2014). Comparison of 12-year-old children with prenatal exposure to cocaine and non-exposed controls on caregiver ratings of executive function. J Youth Adolesc 43: 53–69.
Minozzi S, Amato L, Bellisario C, Ferri M, Davoli M (2013). Maintenance agonist treatments for opiate-dependent pregnant women. Cochrane Database Syst Rev 12: CD006318. Demonstrated that the evidence for suggesting methadone treatment over buprenorphine treatment during pregnancy is actually not thoroughly demonstrated.
Money KM, Stanwood GD (2013). Developmental origins of brain disorders: roles for dopamine. Front Cell Neurosci 7: 260.
Morrow BA, Elsworth JD, Roth RH (2002a). Male rats exposed to cocaine in utero demonstrate elevated expression of Fos in the prefrontal cortex in response to environment. Neuropsychopharmacology 26: 275–285 A terrific rat model of intravenous cocaine furing pregnancy.
Morrow BA, Elsworth JD, Roth RH (2002b). Prenatal cocaine exposure disrupts non-spatial, short-term memory in adolescent and adult male rats. Behav Brain Res 129: 217–223.
Muhammad A, Mychasiuk R, Nakahashi A, Hossain SR, Gibb R, Kolb B (2012). Prenatal nicotine exposure alters neuroanatomical organization of the developing brain. Synapse 66: 950–954.
Mychasiuk R, Muhammad A, Gibb R, Kolb B (2013). Long-term alterations to dendritic morphology and spine density associated with prenatal exposure to nicotine. Brain Res 1499: 53–60.
Nash K, Sheard E, Rovet J, Koren G (2008). Understanding fetal alcohol spectrum disorders (FASDs): toward identification of a behavioral phenotype. ScientificWorldJournal 8: 873–882.
Nash K, Stevens S, Rovet J, Fantus E, Nulman I, Sorbara D et al (2013). Towards identifying a characteristic neuropsychological profile for fetal alcohol spectrum disorders. 1. Analysis of the Motherisk FASD clinic. J Popul Ther Clin Pharmacol 20: e44–e52.
Nassogne MC, Evrard P, Courtoy PJ (1998). Selective direct toxicity of cocaine on fetal mouse neurons. Teratogenic implications of neurite and apoptotic neuronal loss. Ann N Y Acad Sci 846: 51–68.
Navarro M, Rubio P, de Fonseca FR (1995). Behavioural consequences of maternal exposure to natural cannabinoids in rats. Psychopharmacology (Berl) 122: 1–14 Excellent guide on the role of developmental marijuana and resulting behavioral aberrations, primarily in animal models.
Nguyen D, Smith LM, Lagasse LL, Derauf C, Grant P, Shah R et al (2010). Intrauterine growth of infants exposed to prenatal methamphetamine: results from the infant development, environment, and lifestyle study. J Pediatr 157: 337–339.
Oei JL, Kingsbury A, Dhawan A, Burns L, Feller JM, Clews S et al (2012). Amphetamines, the pregnant woman and her children: a review. J Perinatol 32: 737–747 An extensive and detailed review on perinatal AMPH use and its outcomes.
Oliveira MT, Rego AC, Macedo TR, Oliveira CR (2003). Drugs of abuse induce apoptotic features in PC12 cells. Ann N Y Acad Sci 1010: 667–670.
Oliveira MT, Rego AC, Morgadinho MT, Macedo TR, Oliveira CR (2002). Toxic effects of opioid and stimulant drugs on undifferentiated PC12 cells. Ann N Y Acad Sci 965: 487–496.
Ornoy A, Michailevskaya V, Lukashov I, Bar-Hamburger R, Harel S (1996). The developmental outcome of children born to heroin-dependent mothers, raised at home or adopted. Child Abuse Negl 20: 385–396.
Ornoy A, Segal J, Bar-Hamburger R, Greenbaum C (2001). Developmental outcome of school-age children born to mothers with heroin dependency: importance of environmental factors. Dev Med Child Neurol 43: 668–675 Clearly demonstrated that improved environment cannot compensate for all effects seen with prenatal heroin exposure.
Oro AS, Dixon SD (1987). Perinatal cocaine and methamphetamine exposure: maternal and neonatal correlates. J Pediatr 111: 571–578.
Pappalardo-Carter DL, Balaraman S, Sathyan P, Carter ES, Chen WJ, Miranda RC (2013). Suppression and epigenetic regulation of MiR-9 contributes to ethanol teratology: evidence from zebrafish and murine fetal neural stem cell models. Alcohol Clin Exp Res 37: 1657–1667.
Parikh R, Hussain T, Holder G, Bhoyar A, Ewer AK (2011). Maternal methadone therapy increases QTc interval in newborn infants. Arch Dis Child Fetal Neonatal Ed 96: F141–F143.
Paul AP, Pohl-Guimaraes F, Krahe TE, Filgueiras CC, Lantz CL, Colello RJ et al (2010). Overexpression of serum response factor restores ocular dominance plasticity in a model of fetal alcohol spectrum disorders. J Neurosci 30: 2513–2520.
Paul JA, Logan BA, Krishnan R, Heller NA, Morrison DG, Pritham UA et al (2014). Development of auditory event-related potentials in infants prenatally exposed to methadone. Dev Psychobiol 56: 1119–1128.
Pauly JR, Slotkin TA (2008). Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr 97: 1331–1337 Overview of developmental tobacco research that stresses the need for safer alternatives.
Pertschuk LP, Ford DH, Rainford EA (1977). Localization of methadone in fetal rat eye by the immunofluorescence technic. Exp Eye Res 24: 547–552.
Piper BJ, Farelli JD, Meyer JS (2009). Dissociation between serotonin neurotoxicity and brain-derived neurotrophic factor induction following neonatal MDMA exposure in rats. Dev Neurosci 31: 90–94.
Piper BJ, Meyer JS (2006). Increased responsiveness to MDMA in adult rats treated neonatally with MDMA. Neurotoxicol Teratol 28: 95–102 A well-designed and carried out study demonstrating that developmental MDMA administration can induce long-lasting functional abnormalities, including enhanced sensitivity toMDMA itself.
Poland ML, Dombrowski MP, Ager JW, Sokol RJ (1993). Punishing pregnant drug users: enhancing the flight from care. Drug Alcohol Depend 31: 199–203.
Popova S, Stade B, Bekmuradov D, Lange S, Rehm J (2011). What do we know about the economic impact of fetal alcohol spectrum disorder? A systematic literature review. Alcohol Alcohol 46: 490–497.
Porciuncula LO, Sallaberry C, Mioranzza S, Botton PH, Rosemberg DB (2013). The Janus face of caffeine. Neurochem Int 63: 594–609 This is an excellent comprehensive review of the known effects of developmental caffeine exposure.
Powrozek TA, Miller MW (2009). Ethanol affects transforming growth factor beta1-initiated signals: cross-talking pathways in the developing rat cerebral wall. J Neurosci 29: 9521–9533.
Ramamoorthy JD, Ramamoorthy S, Leibach FH, Ganapathy V (1995). Human placental monoamine transporters as targets for amphetamines. Am J Obstet Gynecol 173: 1782–1787.
Rando K, Chaplin TM, Potenza MN, Mayes L, Sinha R (2013). Prenatal cocaine exposure and gray matter volume in adolescent boys and girls: relationship to substance use initiation. Biol Psychiatry 74: 482–489.
Reijneveld SA, Brugman E, Hirasing RA (2002). Excessive infant crying: definitions determine risk groups. Arch Dis Child 87: 43–44.
Ren JQ, Malanga CJ, Tabit E, Kosofsky BE (2004). Neuropathological consequences of prenatal cocaine exposure in the mouse. Int J Dev Neurosci 22: 309–320.
Ricalde AA, Hammer RP Jr (1990). Perinatal opiate treatment delays growth of cortical dendrites. Neurosci Lett 115: 137–143.
Richardson GA, Day NL, Goldschmidt L (1995). Prenatal alcohol, marijuana, and tobacco use: infant mental and motor development. Neurotoxicol Teratol 17: 479–487 Evidence of cognitive deficits in children as young as 9 months of age with prenatal alcohol, marijuana, and tobacco exposure.
Richardson GA, Goldschmidt L, Larkby C, Day NL (2013). Effects of prenatal cocaine exposure on child behavior and growth at 10 years of age. Neurotoxicol Teratol 40: 1–8.
Richardson GA, Goldschmidt L, Willford J (2008). The effects of prenatal cocaine use on infant development. Neurotoxicol Teratol 30: 96–106.
Richardson GA, Ryan C, Willford J, Day NL, Goldschmidt L (2002). Prenatal alcohol and marijuana exposure: effects on neuropsychological outcomes at 10 years. Neurotoxicol Teratol 24: 309–320.
Riley EP, McGee CL (2005). Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 230: 357–365.
Robinson SE (2002). Effects of perinatal buprenorphine and methadone exposures on striatal cholinergic ontogeny. Neurotoxicol Teratol 24: 137–142.
Robinson SE, Maher JR, Wallace MJ, Kunko PM (1997). Perinatal methadone exposure affects dopamine, norepinephrine, and serotonin in the weanling rat. Neurotoxicol Teratol 19: 295–303.
Robinson SE, Mo Q, Maher JR, Wallace MJ, Kunko PM (1996). Perinatal exposure to methadone affects central cholinergic activity in the weanling rat. Drug Alcohol Depend 41: 119–126.
Robinson SE, Wallace MJ (2001). Effect of perinatal buprenorphine exposure on development in the rat. J Pharmacol Exp Ther 298: 797–804.
Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL (2013). Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci 33: 9003–9012.
Rosen TS, Johnson HL (1985). Long-term effects of prenatal methadone maintenance. NIDA Res Monogr 59: 73–83.
Rosenthal DG, Weitzman M, Benowitz NL (2011). Nicotine addiction: mechanisms and consequences. Int J Mental Health 40: 22–38 Thorough discussion of the consequences of developmental tobacco exposure.
Rubio P, Rodriguez de Fonseca F, Munoz RM, Ariznavarreta C, Martin-Calderon JL, Navarro M (1995). Long-term behavioral effects of perinatal exposure to delta 9-tetrahydrocannabinol in rats: possible role of pituitary-adrenal axis. Life Sci 56: 2169–2176.
Ruckinger S, Rzehak P, Chen CM, Sausenthaler S, Koletzko S, Bauer CP et al (2010). Prenatal and postnatal tobacco exposure and behavioral problems in 10-year-old children: results from the GINI-plus prospective birth cohort study. Environ Health Perspect 118: 150–154.
Rudnick G, Wall SC (1992). The molecular mechanism of ‘ecstasy’ [3,4-methylenedioxy-methamphetamine (MDMA)]: serotonin transporters are targets for MDMA-induced serotonin release. Proc Natl Acad Sci USA 89: 1817–1821.
Saadani-Makki F, Frugiere A, Gros F, Gaytan S, Bodineau L (2004). Involvement of adenosinergic A1 systems in the occurrence of respiratory perturbations encountered in newborns following an in utero caffeine exposure. a study on brainstem-spinal cord preparations isolated from newborn rats. Neuroscience 127: 505–518.
Sahir N, Bahi N, Evrard P, Gressens P (2000). Caffeine induces in vivo premature appearance of telencephalic vesicles. Brain Res Dev Brain Res 121: 213–217.
Sahir N, Evrard P, Gressens P (2004). Caffeine induces sonic hedgehog gene expression in cultured astrocytes and neurons. J Mol Neurosci 24: 201–205.
Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P et al (1997). Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology 56: 317–326.
Sanchez ES, Bigbee JW, Fobbs W, Robinson SE, Sato-Bigbee C (2008). Opioid addiction and pregnancy: perinatal exposure to buprenorphine affects myelination in the developing brain. Glia 56: 1017–1027 Provided evidence that suggests a mechanism for altered myelination patterns seen in prenatal opiate exposed neonates.
Santiago SE, Huffman KJ (2013). Prenatal nicotine exposure increases anxiety and modifies sensorimotor integration behaviors in adult female mice. Neurosci Res 79: 41–51.
Sarfi M, Martinsen H, Bakstad B, Roislien J, Waal H (2009). Patterns in sleep-wakefulness in three-month old infants exposed to methadone or buprenorphine. Early Hum Dev 85: 773–778.
Sari Y, Zhou FC (2004). Prenatal alcohol exposure causes long-term serotonin neuron deficit in mice. Alcohol Clin Exp Res 28: 941–948.
Scher MS, Richardson GA, Coble PA, Day NL, Stoffer DS (1988). The effects of prenatal alcohol and marijuana exposure: disturbances in neonatal sleep cycling and arousal. Pediatr Res 24: 101–105.
Schindler SD, Eder H, Ortner R, Rohrmeister K, Langer M, Fischer G (2003). Neonatal outcome following buprenorphine maintenance during conception and throughout pregnancy. Addiction 98: 103–110.
Schneider M (2009). Cannabis use in pregnancy and early life and its consequences: animal models. Eur Arch Psychiatry Clin Neurosci 259: 383–393.
Schneider T, Ilott N, Brolese G, Bizarro L, Asherson PJ, Stolerman IP (2011). Prenatal exposure to nicotine impairs performance of the 5-choice serial reaction time task in adult rats. Neuropsychopharmacology 36: 1114–1125 One of first studies to show prenatal nicotine alters attention in rodents.
Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH et al (2007). Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev 17: 275–297.
Shankaran S, Das A, Bauer CR, Bada HS, Lester BM, Wright LL et al (2011). Prenatal cocaine exposure and small-for-gestational-age status: effects on growth at 6 years of age. Neurotoxicol Teratol 33: 575–581.
Silva CG, Metin C, Fazeli W, Machado NJ, Darmopil S, Launay PS et al (2013). Adenosine receptor antagonists including caffeine alter fetal brain development in mice. Sci Transl Med 5: 197ra104. This exciting paper found that caffeine or another A2A receptor antagonist delayed the migration of specific populations of neurons during brain maturation, increased seizure-susceptibility, and impaired memory.
Silva L, Zhao N, Popp S, Dow-Edwards D (2012). Prenatal tetrahydrocannabinol (THC) alters cognitive function and amphetamine response from weaning to adulthood in the rat. Neurotoxicol Teratol 34: 63–71.
Singer LT, Arendt R, Minnes S, Farkas K, Salvator A (2000). Neurobehavioral outcomes of cocaine-exposed infants. Neurotoxicol Teratol 22: 653–666 Well-designed and implemented study corroborated with previous findings of early effects in cocaine-exposed infants taking into account multiple variables.
Singer LT, Moore DG, Fulton S, Goodwin J, Turner JJ, Min MO et al (2012). Neurobehavioral outcomes of infants exposed to MDMA (Ecstasy) and other recreational drugs during pregnancy. Neurotoxicol Teratol 34: 303–310 Most current study of prenatal MDMA exposure predicting poorer infant mental and motor development at 4 and 12 months of age in a dose-dependent manner.
Skelton MR, Williams MT, Vorhees CV (2008). Developmental effects of 3,4-methylenedioxymethamphetamine: a review. Behav Pharmacol 19: 91–111.
Slamberova R (2012). Drugs in pregnancy: the effects on mother and her progeny. Physiol Res 61: S123–S135.
Slamberova R, Pometlova M, Syllabova L, Mancuskova M (2005). Learning in the place navigation task, not the new-learning task, is altered by prenatal methamphetamine exposure. Dev Brain Res 157: 217–219.
Slotkin TA (2004). Cholinergic systems in brain development and disruption by neurotoxicants: nicotine, environmental tobacco smoke, organophosphates. Toxicol Appl Pharmacol 198: 132–151 Another excellent review by one of the leaders in the nicotine research field.
Slotkin TA (2008). If nicotine is a developmental neurotoxicant in animal studies, dare we recommend nicotine replacement therapy in pregnant women and adolescents? Neurotoxicol Teratol 30: 1–19.
Slotkin TA, Pinkerton KE, Seidler FJ (2006). Perinatal environmental tobacco smoke exposure in rhesus monkeys: critical periods and regional selectivity for effects on brain cell development and lipid peroxidation. Environ Health Perspect 114: 34–39.
Slotkin TA, Weigel SJ, Seidler FJ, Whitmore WL (1982). Toxic effects of maternal methadone administration on cardiac development in the neonatal rat: potential participation of altered polyamine levels in growth impairment. Toxicol Lett 14: 131–135 Provided a potential mechanism for transient cardiac abnormalities seen in patients.
Smith AM, Fried PA, Hogan MJ, Cameron I (2004). Effects of prenatal marijuana on response inhibition: an fMRI study of young adults. Neurotoxicol Teratol 26: 533–542 One of first studies to demonstrate that the effects of prenatal marijuana last well into adulthood.
Smith AM, Fried PA, Hogan MJ, Cameron I (2006a). Effects of prenatal marijuana on visuospatial working memory: an fMRI study in young adults. Neurotoxicol Teratol 28: 286–295.
Smith L, Yonekura ML, Wallace T, Berman N, Kuo J, Berkowitz C (2003). Effects of prenatal methamphetamine exposure on fetal growth and drug withdrawal symptoms in infants born at term. J Dev Behav Pediatr 24: 17–23.
Smith LM, LaGasse LL, Derauf C, Grant P, Shah R, Arria A et al (2006b). The infant development, environment, and lifestyle study: effects of prenatal methamphetamine exposure, polydrug exposure, and poverty on intrauterine growth. Pediatrics 118: 1149–1156.
Smith LM, LaGasse LL, Derauf C, Newman E, Shah R, Haning W et al (2011). Motor and cognitive outcomes through three years of age in children exposed to prenatal methamphetamine. Neurotoxicol Teratol 33: 176–184.
Soellner DE, Grandys T, Nunez JL (2009). Chronic prenatal caffeine exposure impairs novel object recognition and radial arm maze behaviors in adult rats. Behav Brain Res 205: 191–199.
Soepatmi S (1994). Developmental outcomes of children of mothers dependent on heroin or heroin/methadone during pregnancy. Acta Paediatr Suppl 404: 36–39.
Sorenson CA, Raskin LA, Suh Y (1991). The effects of prenatal nicotine on radial-arm maze performance in rats. Pharmacol Biochem Behav 40: 991–993.
Spano MS, Ellgren M, Wang X, Hurd YL (2007). Prenatal cannabis exposure increases heroin seeking with allostatic changes in limbic enkephalin systems in adulthood. Biol Psychiatry 61: 554–563.
Spear LP (2014). Adolescents and alcohol: acute sensitivities, enhanced intake, and later consequences. Neurotoxicol Teratol 41C: 51–59 An up-to-date comprehensive review of critical brain changes during adolescence and how alcohol induces behavioral, cognitive, and neural alterations that are highly specific, replicable, persistent and dependent on the timing of the exposure during maturation.
Spear LP, Varlinskaya EI (2005). Adolescence. Alcohol sensitivity, tolerance, and intake. Recent Dev Alcohol 17: 143–159.
Stanwood GD, Levitt P (2003). Repeated i.v. cocaine exposure produces long-lasting behavioral sensitization in pregnant adults, but behavioral tolerance in their offspring. Neuroscience 122: 579–583.
Stanwood GD, Levitt P (2004). Drug exposure early in life: functional repercussions of changing neuropharmacology during sensitive periods of brain development. Curr Opin Pharmacol 4: 65–71.
Stanwood GD, Parlaman JP, Levitt P (2005). Anatomical abnormalities in dopaminoceptive regions of the cerebral cortex of dopamine D1 receptor mutant mice. J Comp Neurol 487: 270–282.
Stanwood GD, Washington RA, Levitt P (2001a). Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate cortex. Cereb Cortex 11: 430–440.
Stanwood GD, Washington RA, Shumsky JS, Levitt P (2001b). Prenatal cocaine exposure produces consistent developmental alterations in dopamine-rich regions of the cerebral cortex. Neuroscience 106: 5–14.
Steingart RA, Abu-Roumi M, Newman ME, Silverman WF, Slotkin TA, Yanai J (2000a). Neurobehavioral damage to cholinergic systems caused by prenatal exposure to heroin or phenobarbital: cellular mechanisms and the reversal of deficits by neural grafts. Brain Res Dev Brain Res 122: 125–133.
Steingart RA, Silverman WF, Barron S, Slotkin TA, Awad Y, Yanai J (2000b). Neural grafting reverses prenatal drug-induced alterations in hippocampal PKC and related behavioral deficits. Brain Res Dev Brain Res 125: 9–19 Provided evidence for the functional significance of hippocampal cholinergic deficits in models of prenatal opiate exposure.
Stene-Larsen K, Borge AI, Vollrath ME (2009). Maternal smoking in pregnancy and externalizing behavior in 18-month-old children: results from a population-based prospective study. J Am Acad Child Adolesc Psychiatry 48: 283–289.
Stroud LR, Paster RL, Goodwin MS, Shenassa E, Buka S, Niaura R et al (2009a). Maternal smoking during pregnancy and neonatal behavior: a large-scale community study. Pediatrics 123: e842–e848.
Stroud LR, Paster RL, Papandonatos GD, Niaura R, Salisbury AL, Battle C et al (2009b). Maternal smoking during pregnancy and newborn neurobehavior: effects at 10 to 27 days. J Pediatr 154: 10–16.
Substance Abuse and Mental Health Services Administration (2008) The NSDUH Report—Alcohol Use among Pregnant Women and Recent Mothers: 2002 to 2007. Office of Applied Studies: Rockville, MD.
Substance Abuse and Mental Health Services Administration (2009) The NSDUH Report: Substance Use among Women During Pregnancy and Following Childbirth. Office of Applied Studies: Rockville, MD.
Substance Abuse and Mental Health Services Administration (2013) Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings. Administration, S. A. a. M. H. S: Rockville, MD.
Sundelin Wahlsten V, Sarman I (2013). Neurobehavioural development of preschool-age children born to addicted mothers given opiate maintenance treatment with buprenorphine during pregnancy. Acta Paediatr 102: 544–549.
Svensson AL, Bucht N, Hallberg M, Nyberg F (2008). Reversal of opiate-induced apoptosis by human recombinant growth hormone in murine foetus primary hippocampal neuronal cell cultures. Proc Natl Acad Sci USA 105: 7304–7308.
Tempel A (1991). Visualization of mu opiate receptor downregulation following morphine treatment in neonatal rat brain. Brain Res Dev Brain Res 64: 19–26.
Tennes K, Avitable N, Blackard C, Boyles C, Hassoun B, Holmes L et al (1985). Marijuana: prenatal and postnatal exposure in the human. NIDA Res Monogr 59: 48–60.
Thompson BL, Levitt P, Stanwood GD (2005). Prenatal cocaine exposure specifically alters spontaneous alternation behavior. Behav Brain Res 164: 107–116.
Thompson BL, Levitt P, Stanwood GD (2009a). Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat Rev Neurosci 10: 303–312.
Thompson VB, Heiman J, Chambers JB, Benoit SC, Buesing WR, Norman MK et al (2009b). Long-term behavioral consequences of prenatal MDMA exposure. Physiol Behav 96: 593–601.
Thompson VB, Koprich JB, Chen EY, Kordower JH, Terpstra BT, Lipton JW (2012). Prenatal exposure to MDMA alters noradrenergic neurodevelopment in the rat. Neurotoxicol Teratol 34: 206–213.
Tong VT, Dietz PM, Morrow B, D'Angelo DV, Farr SL, Rockhill KM et al (2013). Trends in smoking before, during, and after pregnancy—Pregnancy Risk Assessment Monitoring System, United States, 40 sites, 2000-2010. MMWR Surveill Summ 62: 1–19.
Treit S, Lebel C, Baugh L, Rasmussen C, Andrew G, Beaulieu C (2013). Longitudinal MRI reveals altered trajectory of brain development during childhood and adolescence in fetal alcohol spectrum disorders. J Neurosci 33: 10098–10109.
Trezza V, Campolongo P, Cassano T, Macheda T, Dipasquale P, Carratu MR et al (2008a). Effects of perinatal exposure to delta-9-tetrahydrocannabinol on the emotional reactivity of the offspring: a longitudinal behavioral study in Wistar rats. Psychopharmacology (Berl) 198: 529–537.
Trezza V, Cuomo V, Vanderschuren LJ (2008b). Cannabis and the developing brain: insights from behavior. Eur J Pharmacol 585: 441–452.
Tronick EZ, Messinger DS, Weinberg MK, Lester BM, Lagasse L, Seifer R et al (2005). Cocaine exposure is associated with subtle compromises of infants' and mothers' social-emotional behavior and dyadic features of their interaction in the face-to-face still-face paradigm. Dev Psychol 41: 711–722.
Tsang D, Ho KP, Wen HL (1986). Effect of maternal methadone administration on the development of multiple forms of monoamine oxidase in rat brain and liver. Brain Res 391: 187–192.
Twomey J, LaGasse L, Derauf C, Newman E, Shah R, Smith L et al (2013). Prenatal methamphetamine exposure, home environment, and primary caregiver risk factors predict child behavioral problems at 5 years. Am J Orthopsychiatry 83: 64–72 Study invested the possible association between prenatal methamphetamine exposure and child behavioral problems at 5 years and closely examining home environment and primary caregiver risk factors.
U.S. Department of Health and Human Services (2010) How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General.. Atlanta, GA.
Ungerer M, Knezovich J, Ramsay M (2013). In utero alcohol exposure, epigenetic changes, and their consequences. Alcohol Res 35: 37–46.
UNODC (2013) World Drug Report 2013. United Nations Publication.
Vaglenova J, Parameshwaran K, Suppiramaniam V, Breese CR, Pandiella N, Birru S (2008). Long-lasting teratogenic effects of nicotine on cognition: gender specificity and role of AMPA receptor function. Neurobiol Learn Mem 90: 527–536.
Valenzuela CF, Morton RA, Diaz MR, Topper L (2012). Does moderate drinking harm the fetal brain? Insights from animal models. Trends Neurosci 35: 284–292.
van Baar A (1990). Development of infants of drug dependent mothers. J Child Psychol Psychiatry 31: 911–920.
van Baar A, de Graaff BM (1994). Cognitive development at preschool-age of infants of drug-dependent mothers. Dev Med Child Neurol 36: 1063–1075.
van Baar AL, Soepatmi S, Gunning WB, Akkerhuis GW (1994). Development after prenatal exposure to cocaine, heroin and methadone. Acta Paediatr Suppl 404: 40–46.
van Tonningen-van Driel MM, Garbis-Berkvens JM, Reuvers-Lodewijks WE (1999). [Pregnancy outcome after ecstasy use; 43 cases followed by the Teratology Information Service of the National Institute for Public Health and Environment (RIVM)]. Ned Tijdschr Geneeskd 143: 27–31.
Vargas GC, Pildes RS, Vidyasagar D, Keith LG (1975). Effect of maternal heroin addiction on 67 liveborn neonates. Withdrawal symptoms, small body size, and small head circumference were frequent findings. Clin Pediatr (Phila) 14: 751–753.
Varlinskaya EI, Spear LP (2002). Acute effects of ethanol on social behavior of adolescent and adult rats: role of familiarity of the test situation. Alcohol Clin Exp Res 26: 1502–1511.
Vassoler FM, White SL, Schmidt HD, Sadri-Vakili G, Pierce RC (2013). Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci 16: 42–47.
Vathy I, Katay L (1992). Effects of prenatal morphine on adult sexual behavior and brain catecholamines in rats. Brain Res Dev Brain Res 68: 125–131.
Villarreal DM, Derrick B, Vathy I (2008). Prenatal morphine exposure attenuates the maintenance of late LTP in lateral perforant path projections to the dentate gyrus and the CA3 region in vivo. J Neurophysiol 99: 1235–1242.
Vorhees CV, Inman-Wood SL, Morford LL, Broening HW, Fukumura M, Moran MS (2000). Adult learning deficits after neonatal exposure to D-methamphetamine: selective effects on spatial navigation and memory. J Neurosci 20: 4732–4739.
Vorhees CV, Reed TM, Skelton MR, Williams MT (2004). Exposure to 3,4-methylenedioxymethamphetamine (MDMA) on postnatal days 11-20 induces reference but not working memory deficits in the Morris water maze in rats: implications of prior learning. Int J Dev Neurosci 22: 247–259.
Wakschlag LS, Leventhal BL, Pine DS, Pickett KE, Carter AS (2006a). Elucidating early mechanisms of developmental psychopathology: the case of prenatal smoking and disruptive behavior. Child Dev 77: 893–906.
Wakschlag LS, Pickett KE, Kasza KE, Loeber R (2006b). Is prenatal smoking associated with a developmental pattern of conduct problems in young boys? J Am Acad Child Adolesc Psychiatry 45: 461–467.
Walhovd KB, Watts R, Amlien I, Woodward LJ (2012). Neural tract development of infants born to methadone-maintained mothers. Pediatr Neurol 47: 1–6 Demonstrated novel finding of altered development of myelination patterns in prenatal opiate exposed neonates.
Wang X, Dow-Edwards D, Anderson V, Minkoff H, Hurd YL (2004). In utero marijuana exposure associated with abnormal amygdala dopamine D2 gene expression in the human fetus. Biol Psychiatry 56: 909–915.
Wang X, Dow-Edwards D, Keller E, Hurd YL (2003). Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience 118: 681–694.
Wang Y, Han TZ (2009). Prenatal exposure to heroin in mice elicits memory deficits that can be attributed to neuronal apoptosis. Neuroscience 160: 330–338.
Warren KR, Li TK (2005). Genetic polymorphisms: impact on the risk of fetal alcohol spectrum disorders. Birth Defects Res A Clin Mol Teratol 73: 195–203.
Weitzman M, Gortmaker S, Sobol A (1992). Maternal smoking and behavior problems of children. Pediatrics 90: 342–349.
Welle-Strand GK, Skurtveit S, Jones HE, Waal H, Bakstad B, Bjarko L et al (2013). Neonatal outcomes following in utero exposure to methadone or buprenorphine: a National Cohort Study of opioid-agonist treatment of Pregnant Women in Norway from 1996 to 2009. Drug Alcohol Depend 127: 200–206.
Willford JA, Richardson GA, Leech SL, Day NL (2004). Verbal and visuospatial learning and memory function in children with moderate prenatal alcohol exposure. Alcohol Clin Exp Res 28: 497–507.
Williams MT, Blankenmeyer TL, Schaefer TL, Brown CA, Gudelsky GA, Vorhees CV (2003a). Long-term effects of neonatal methamphetamine exposure in rats on spatial learning in the Barnes maze and on cliff avoidance, corticosterone release, and neurotoxicity in adulthood. Brain Res Dev Brain Res 147: 163–175.
Williams MT, Moran MS, Vorhees CV (2003b). Refining the critical period for methamphetamine-induced spatial deficits in the Morris water maze. Psychopharmacology (Berl) 168: 329–338.
Williams MT, Morford LL, Wood SL, Rock SL, McCrea AE, Fukumura M et al (2003c). Developmental 3,4-methylenedioxymethamphetamine (MDMA) impairs sequential and spatial but not cued learning independent of growth, litter effects or injection stress. Brain Res 968: 89–101.
Williams MT, Morford LL, Wood SL, Wallace TL, Fukumura M, Broening HW et al (2003d). Developmental D-methamphetamine treatment selectively induces spatial navigation impairments in reference memory in the Morris water maze while sparing working memory. Synapse 48: 138–148.
Williams MT, Vorhees CV, Boon F, Saber AJ, Cain DP (2002). Methamphetamine exposure from postnatal day 11 to 20 causes impairments in both behavioral strategies and spatial learning in adult rats. Brain Res 958: 312–321 Data suggest that sensorimotor deficits do not account for the long-term spatial learning and memory deficits in animals exposed neonatally to methamphetamine.
Wilson GS, Desmond MM, Wait RB (1981). Follow-up of methadone-treated and untreated narcotic-dependent women and their infants: health, developmental, and social implications. J Pediatr 98: 716–722.
Winslow JT, Insel TR (1990). Serotonergic modulation of rat pup ultrasonic vocal development: studies with 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther 254: 212–220.
Witter FR, Niebyl JR (1990). Marijuana use in pregnancy and pregnancy outcome. Am J Perinatol 7: 36–38.
Wittert G, Hope P, Pyle D (1996). Tissue distribution of opioid receptor gene expression in the rat. Biochem Biophys Res Commun 218: 877–881.
Wong CS, Lee YJ, Chiang YC, Fan LW, Ho IK, Tien LT (2014). Effect of prenatal methadone on reinstated behavioral sensitization induced by methamphetamine in adolescent rats. Behav Brain Res 258: 160–165.
Xu D, Wu Y, Liu F, Liu YS, Shen L, Lei YY et al (2012a). A hypothalamic-pituitary-adrenal axis-associated neuroendocrine metabolic programmed alteration in offspring rats of IUGR induced by prenatal caffeine ingestion. Toxicol Appl Pharmacol 264: 395–403.
Xu D, Zhang B, Liang G, Ping J, Kou H, Li X et al (2012b). Caffeine-induced activated glucocorticoid metabolism in the hippocampus causes hypothalamic-pituitary-adrenal axis inhibition in fetal rats. PLoS One 7: e44497.
Yanai J, Pick CG, Rogel-Fuchs Y, Zahalka EA (1992). Alterations in hippocampal cholinergic receptors and hippocampal behaviors after early exposure to nicotine. Brain Res Bull 29: 363–368.
Yang SN, Liu CA, Chung MY, Huang HC, Yeh GC, Wong CS et al (2006). Alterations of postsynaptic density proteins in the hippocampus of rat offspring from the morphine-addicted mother: beneficial effect of dextromethorphan. Hippocampus 16: 521–530.
Yaniv SP, Naor Z, Yanai J (2004). Prenatal heroin exposure alters cholinergic receptor stimulated activation of the PKCbetaII and PKCgamma isoforms. Brain Res Bull 63: 339–349.
Zabaneh R, Smith LM, LaGasse LL, Derauf C, Newman E, Shah R et al (2012). The effects of prenatal methamphetamine exposure on childhood growth patterns from birth to 3 years of age. Am J Perinatol 29: 203–210.
Zagon IS, Gibo DM, McLaughlin PJ (1992). Ontogeny of zeta (zeta), the opioid growth factor receptor, in the rat brain. Brain Res 596: 149–156.
Zagon IS, McLaughlin PJ (1977a). Effect of chronic maternal methadone exposure on perinatal development. Biol Neonate 31: 271–282.
Zagon IS, McLaughlin PJ (1977b). Effects of chronic morphine administration on pregnant rats and their offspring. Pharmacology 15: 302–310.
Zagon IS, McLaughlin PJ (1977c). Morphine and brain growth retardation in the rat. Pharmacology 15: 276–282.
Zagon IS, McLaughlin PJ (1978). Perinatal methadone exposure and its influence on the behavioral ontogeny of rats. Pharmacol Biochem Behav 9: 665–672.
Zagon IS, McLaughlin PJ (1979). Motor activity and learning ability in rats perinatally exposed to methadone. NIDA Res Monogr 27: 121–127.
Zagon IS, McLaughlin PJ (1982). Neuronal cell deficits following exposure to methadone in rats. Experientia 38: 1214–1216.
Zagon IS, McLaughlin PJ (1983). Somatic and neurobiological alterations in the progeny of female rats treated with methadone prior to mating. NIDA Res Monogr 43: 184–189.
Zagon IS, McLaughlin PJ (1984). Naltrexone modulates body and brain development in rats: a role for endogenous opioid systems in growth. Life Sci 35: 2057–2064.
Zagon IS, McLaughlin PJ (1987). Endogenous opioid systems regulate cell proliferation in the developing rat brain. Brain Res 412: 68–72 Provided some of the initial evidence that endogenous opiates function to inhibit neuronal proliferation.
Zagon IS, McLaughlin PJ, Thompson CI (1979). Learning ability in adult female rats perinatally exposed to methadone. Pharmacol Biochem Behav 10: 889–894.
Zagon IS, Tobias SW, Hytrek SD, McLaughlin PJ (1998). Opioid receptor blockade throughout prenatal life confers long-term insensitivity to morphine and alters mu opioid receptors. Pharmacol Biochem Behav 59: 201–207.
Zagon IS, Wu Y, McLaughlin PJ (1999). Opioid growth factor and organ development in rat and human embryos. Brain Res 839: 313–322.
Zhen X, Torres C, Wang HY, Friedman E (2001). Prenatal exposure to cocaine disrupts D1A dopamine receptor function via selective inhibition of protein phosphatase 1 pathway in rabbit frontal cortex. J Neurosci 21: 9160–9167.
Zhou FC, Sari Y, Powrozek T, Goodlett CR, Li TK (2003). Moderate alcohol exposure compromises neural tube midline development in prenatal brain. Brain Res Dev Brain Res 144: 43–55.
Zhou FC, Sari Y, Powrozek TA (2005). Fetal alcohol exposure reduces serotonin innervation and compromises development of the forebrain along the serotonergic pathway. Alcohol Clin Exp Res 29: 141–149.
Zhu J, Lee KP, Spencer TJ, Biederman J, Bhide PG (2014). Transgenerational transmission of hyperactivity in a mouse model of ADHD. J Neurosci 34: 2768–2773 The authors stunningly demonstrated transgenerational transmission of prenatal nicotine-induced hyperactivity in mice via the maternal but not the paternal line of descent, suggesting that transgenerational transmission may propagate environmentally induced ADHD phenotypes.
Zhu J, Zhang X, Xu Y, Spencer TJ, Biederman J, Bhide PG (2012). Prenatal nicotine exposure mouse model showing hyperactivity, reduced cingulate cortex volume, reduced dopamine turnover, and responsiveness to oral methylphenidate treatment. J Neurosci 32: 9410–9418.
Zmitrovich AC, Brake SC, Liu PY, Hamowy AS, Hutchings DE (1994). Prenatal administration of methadone in the rat: acoustic startle amplitude and the rest-activity cycle at 30 days of age. Neurotoxicol Teratol 16: 251–255.
Acknowledgements
We are grateful to innumerable colleagues who have helped to shape our views on these topics, including Drs Pat Levitt, Jeremy Veenstra-Vanderweele, Gale Richardson, Charles Vorhees, Randy Blakely, Barbara Thompson, and Pradeep Bhide. We also thank the reviewers who provided substantial feedback and very helpful suggestions on an earlier draft of this review. We are currently supported by RO1MH086629 (GDS), the Vanderbilt Conte Center (GDS and EJR), the McManus Foundation (DLG), and the Vanderbilt Brain Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ross, E., Graham, D., Money, K. et al. Developmental Consequences of Fetal Exposure to Drugs: What We Know and What We Still Must Learn. Neuropsychopharmacol 40, 61–87 (2015). https://doi.org/10.1038/npp.2014.147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.147
This article is cited by
-
Molecular signatures of angiogenesis inhibitors: a single-embryo untargeted metabolomics approach in zebrafish
Archives of Toxicology (2024)
-
Early postnatal microglial ablation in the Ccdc39 mouse model reveals adverse effects on brain development and in neonatal hydrocephalus
Fluids and Barriers of the CNS (2023)
-
In vitro modeling of the human dopaminergic system using spatially arranged ventral midbrain–striatum–cortex assembloids
Nature Methods (2023)
-
Whole transcriptome analysis in offspring whose fathers were exposed to a developmental insult: a novel avian model
Scientific Reports (2023)
-
The Genetically Informed Neurobiology of Addiction (GINA) model
Nature Reviews Neuroscience (2023)
