
Abstract
Fragile X syndrome (FXS) is an inherited neurodevelopmental disease caused by loss of function of the fragile X mental retardation protein (FMRP). In the absence of FMRP, signaling through group 1 metabotropic glutamate receptors is elevated and insensitive to stimulation, which may underlie many of the neurological and neuropsychiatric features of FXS. Treatment of FXS animal models with negative allosteric modulators of these receptors and preliminary clinical trials in human patients support the hypothesis that metabotropic glutamate receptor signaling is a valuable therapeutic target in FXS. However, recent research has also shown that FMRP may regulate diverse aspects of neuronal signaling downstream of several cell surface receptors, suggesting a possible new route to more direct disease-targeted therapies. Here, we summarize promising recent advances in basic research identifying and testing novel therapeutic strategies in FXS models, and evaluate their potential therapeutic benefits. We provide an overview of recent and ongoing clinical trials motivated by some of these findings, and discuss the challenges for both basic science and clinical applications in the continued development of effective disease mechanism-targeted therapies for FXS.
Similar content being viewed by others

INTRODUCTION
A majority of intellectual disabilities are idiopathic. Therefore, therapeutic strategies to treat these disorders are not usually designed to target the underlying pathological mechanisms. Rather, treatments are mostly limited to behavioral interventions or drug therapies that ameliorate comorbid symptoms, such as anxiety, aggression, or epilepsy (Davis et al, 2008; Depositario-Cabacar and Zelleke, 2010; Brosnan and Healy, 2011). Fragile X Syndrome (FXS) is the most common inherited form of intellectual disability (ID) and the most common known monogenic cause of autism spectrum disorders (ASD). Research analyzing the underlying molecular mechanisms has led to the discovery of several promising targets for disease-mitigating pharmacological therapies, which might also be useful in treating other cognitive disorders and ASDs (Wang et al, 2010b).
FXS is caused by a CGG trinucleotide expansion in the 5′ untranslated region of the X-chromosomal fragile X mental retardation 1 gene (FMR1) (Verkerk et al, 1991). This mutation leads to hypermethylation of the surrounding nucleotide sequences and transcriptional silencing of the FMR1 gene (Pieretti et al, 1991). As a consequence, the encoded fragile X mental retardation protein (FMRP) is not expressed. The length of the trinucleotide expansion in FMR1 determines disease expression; alleles containing ⩽44 repeats are considered normal, gray zone alleles have 45–54 repeats, premutation alleles contain between 55 and 200 repeats, and alleles with ⩾200 repeats are considered a full mutation (Maddalena et al, 2001). Only the full mutation causes silencing of the gene and thus FXS. However, premutation can lead to fragile X-associated premature ovarian insufficiency (FXPOI) in female carriers (Sherman, 2000) and an adult-onset neurodegenerative disease, the fragile X-associated tremor/ataxia syndrome (FXTAS) (Berry-Kravis et al, 2007). Apart from the usual CGG repeat expansion resulting in FXS, a missense mutation within FMRP was found in a severe case of FXS (De Boulle et al, 1993). Moreover, two recent studies identified several mutations within the open reading frame and the promoter region of the FMR1 gene in patients with neurodevelopmental diseases and disorders similar to FXS, suggesting that dysregulation or dysfunction of FMRP is the cause of FXS-like symptoms (Collins et al, 2010a, 2010b).
In this review, we summarize and evaluate advances in recent research elucidating the molecular functions of FMRP. We assess the potential of these findings to lead to therapeutic strategies for FXS or other disorders caused by FMR1 loss-of-function mutations. We discuss current clinical trials targeting some of the pathological mechanisms caused by the absence of FMRP.
FRAGILE X CLINICAL PHENOTYPE
Individuals with a fragile X full mutation and FXS display characteristic physical features, ID, dysfunction in multiple behavioral domains, and specific medical problems. All aspects of the phenotype are more evident in males than females, as females express FMRP from the normal X chromosome, the amount of which depends on X inactivation ratios. Physical features and associated medical problems (Berry-Kravis et al, 2002; Hagerman et al, 2009) are variably present and include: macroorchidism (present in most adult males), prominent ears, macrocephaly, long face, high arched palate, hyperextensible joints, soft skin, recurrent otitis media and sinusitis, gastroesophageal reflux, strabismus and farsightedness (Maino et al, 1991), hyperphagia and obesity (Raspa et al, 2010; Utari et al, 2010), mitral valve prolapse and valvular insufficiency (particularly in adults), pes planus (flat feet), joint dislocations, scoliosis, disrupted sleep patterns (Kronk et al, 2010), and obstructive sleep apnea. Seizures occur in ∼15% of males and 6–8% of females with FXS, and most commonly are partial complex (Musumeci et al, 1999; Berry-Kravis, 2002; Berry-Kravis et al, 2010), have onset between age 4 and 10 years, and resolve during childhood, although presence of seizures appears to be an indicator of an increased risk for autism in FXS (Berry-Kravis, 2002; Berry-Kravis et al, 2010). Although important to manage when present, the medical and physical problems are mild, and cognitive and behavioral impairments are the aspects of the phenotype that result in substantial functional limitations.
Cognitive/Developmental Phenotype
Young children with FXS are typically identified because of presentation with developmental (particularly speech) delays and/or hypotonia (Berry-Kravis et al, 2002). Males with a completely methylated full mutation usually display mild-to-moderate ID (average IQ 40–50, mental age 5 to 6 years for adults) with adaptive/achievement skills typically higher than IQ would predict (Berry-Kravis et al, 2002; Hagerman, 2002; Hagerman et al, 2009). IQ can be higher in young boys and decline during mid-childhood in the absence of true regression, because of poor acquisition of complex processing skills relative to typically developing children (Berry-Kravis et al, 2002; Hagerman, 2002). Females with the full mutation often present with executive and non-verbal learning disabilities, although the average IQ is about 80 and ∼25% have ID (de Vries et al, 1996). Males and females with FXS have a qualitatively similar cognitive profile that is quantitatively less severe in females, with strengths in receptive vocabulary, grammatical structure, visual memory, simultaneous processing, experiential learning, and imitation, and weaknesses in auditory processing, sequential processing, mathematical and quantity skills, abstraction, visuospatial and constructional ability, working memory, executive function and attention, and coordination and praxis (Berry-Kravis et al, 2002; Braden, 2002). Knowledge of the characteristic cognitive profile in FXS allows educational and therapy programming to be tailored to enhance learning by capitalizing on strengths and minimizing weaknesses (Braden, 2002).
Behavioral Phenotype
Males with FXS have characteristic behavioral features including hyperactivity, impulsivity, attention problems, generalized anxiety with phobias and multiple specific domains of anxiety (de Vries et al, 1996; Sullivan et al, 2007; Hagerman et al, 2009; Farzin et al, 2011), mood lability, and autistic features such as poor eye contact, shyness, self-talk, hand-flapping, hand-biting, hyperarousal to sensory stimuli, and substantial perseverative language and behavior (Berry-Kravis and Potanos, 2004; Hagerman et al, 2009; Wang et al, 2010b). A subset of these problems are common in females, including attention problems, impulsivity, shyness, selective mutism, specific phobias, social anxiety, and social deficits, often occurring even when the IQ is in the normal range (de Vries et al, 1996; Hagerman et al, 2009; Wang et al, 2010b). Individuals with FXS demonstrate an enhanced sympathetic response to sensory stimuli, as measured by electrodermal responses (Miller et al, 1999), heart rate variability (Boccia and Roberts, 2000), and pupillary responses (Farzin et al, 2009), and abnormal sensory gating in prepulse inhibition studies (Hessl et al, 2009). Aggression occurs in at least 30–50% of males, most often in adolescence (Hessl et al, 2008). Many boys with FXS (50–90%) display autistic behavioral patterns, particularly avoidant and anxious behaviors, and ∼18–36% meet full criteria for Autistic Disorder, whereas ∼43–67% of males and 20–23% of females with FXS have an ASD (Wang et al, 2010b). The patterns of social behavior in FXS often differ from typical autism even in individuals with FXS who meet criteria for ASD: individuals with FXS often have strong social interest but high levels of social anxiety. The behaviors in FXS often limit performance, and supportive treatment with behavioral therapy and medications targeted to the most severe behavioral symptoms are commonly utilized to improve functioning.
FMRP: A KEY REGULATOR OF NEURONAL FUNCTION
A major challenge in basic science is to understand how the loss of FMRP results in the behavioral and cognitive deficits in FXS. FMRP is an mRNA-binding protein that plays a role in various aspects of activity-dependent mRNA metabolism in neurons, such as mRNA transport (Dictenberg et al, 2008; Estes et al, 2008), stability (Zalfa et al, 2007), and regulation of mRNA translation (see, eg, Zalfa et al, 2003; Muddashetty et al, 2007; Bechara et al, 2009). Here, we will focus on the role of FMRP in translational control, as this is the so far best-studied function of FMRP in RNA metabolism.
The molecular mechanisms whereby FMRP regulates basal and stimulus-induced translation are not fully understood and are controversially discussed in the field. FMRP was shown to repress translation of individual target mRNAs as well as general protein synthesis (Zalfa et al, 2003; Lu et al, 2004; Dolen et al, 2007; Muddashetty et al, 2007; Gross et al, 2010; Osterweil et al, 2010). On the other hand, recent work suggests that FMRP can also act as a translational activator for specific target mRNAs (Bechara et al, 2009; Fahling et al, 2009; Gross et al, 2011). FMRP has been proposed to regulate not only translation initiation (Zalfa et al, 2006; Napoli et al, 2008), but also translation elongation (Darnell et al, 2005). In addition, FMRP is associated with the RISC complex (Jin et al, 2004), implying a role in microRNA-mediated translational control. In fact, recent studies indicate that FMRP regulates translation of certain cargo mRNAs via specific microRNAs targeted to their 3′UTR sequences (Edbauer et al, 2010; Muddashetty et al, 2011). Future studies will have to address whether and how FMRP regulates different steps during mRNA translation and if this regulation varies for specific targets.
FMRP contains several RNA-binding domains: two KH domains, a C-terminal RGG box, and a more recently discovered N-terminal mRNA-binding motif (reviewed in Bassell and Warren, 2008). Although the RGG box was shown to associate with mRNAs carrying a G-quartet in their 3′UTR (Darnell et al, 2001), no physiological mRNA targets for the KH domains have been discovered so far. An in vitro study identified a tertiary mRNA structure, named the kissing complex, which binds to the second KH (KH2) domain of FMRP (Darnell et al, 2005). This study also suggested that the interaction of the KH2 domain with kissing complex harboring RNAs mediates the association of FMRP with polysomes. Of note, a missense mutation within the KH2 domain, which was found in a severe case of FXS (De Boulle et al, 1993), leads to loss of polysomal association of FMRP (Feng et al, 1997).
FMRP is involved in and affected by many activity-dependent neuronal mechanisms. In vitro screens have suggested that FMRP might associate with up to 4% of all mRNAs present in the brain (Brown et al, 2001; Miyashiro et al, 2003), and the identified mRNA targets encode proteins important for various cellular mechanisms, such as cytoskeletal regulation, AMPA receptor trafficking, synaptic structure and composition, and synaptic signal transmission (Zalfa et al, 2003; Lu et al, 2004; Muddashetty et al, 2007; Schutt et al, 2009; Strumbos et al, 2010). FMRP is not only a multifaceted regulator of numerous target mRNAs, but is also itself activity-regulated at multiple steps, such as FMRP synthesis at synapses (Weiler et al, 1997), protein and mRNA localization in dendrites and at synapses (Antar et al, 2004), and posttranslational modifications and protein stability (reviewed in Bassell and Warren, 2008; De Rubeis and Bagni, 2010; Pfeiffer and Huber, 2009). In summary, these studies indicate that FMRP is tightly regulated in response to neuronal stimuli, and plays diverse roles in neuronal activity-dependent mechanisms of RNA metabolism. This complexity of functions and regulatory mechanisms, together with the variety of putative mRNA targets, suggests that FMRP is a key regulator of neuronal and synaptic development and plasticity, and might explain the various neurological problems associated with FXS. Here, we will focus on the emerging role of FMRP to regulate neuronal signal transduction and the promising therapeutic strategies that are arising from this function.
ANIMAL MODELS OF FXS
The knowledge about neuronal and molecular mechanisms that underlie FXS has been greatly advanced by the generation and analysis of animal models. The first FXS animal model developed was the Fmr1 knockout (KO) mouse (The Dutch-Belgian Fragile et al, 1994), followed by Drosophila FXS models with a deleted or mutated dfmr1 gene (Zhang et al, 2001; Morales et al, 2002), and zebrafish FXS models, in which fmr1 expression was knocked down with antisense morpholinos or the fmr1 gene was deleted by genetic knockout (Tucker et al, 2006; den Broeder et al, 2009). These transgenic or knockout animals do not carry the trinucleotide expansion, but do have functional deletions of FMRP. Although they are, therefore, not perfect models of the human disease, they have helped tremendously to reveal the molecular mechanisms underlying FXS. They recapitulate several symptoms observed in human patients, and the phenotypic presentation is comparably broad, with defects in neuronal development, dendritic spine morphology, synaptic plasticity, pain processing, and behavior.
The characterization of the FXS mouse model has been partially difficult because most phenotypes have rather small effect sizes and are sensitive to genetic background and experimental settings. Thus, several previously described molecular, cell biological, or behavioral phenotypes were difficult to reproduce in different laboratories, and are controversially discussed in the field (see below). Nevertheless, FXS animal models have been proven helpful to identify, develop, and test potential therapeutic strategies to treat FXS. Here, we will primarily focus on (1) robust phenotypes that are shown to be useful to test the effects of specific drug treatments, and (2) recently discovered novel phenotypes that might become helpful assessment tools for FXS-targeted therapeutic strategies in the future.
Abnormal Dendritic Spine Morphology in FXS
Fragile X, like many other neurological diseases (Penzes et al, 2011), is characterized by abnormal dendritic spine morphology. Observations in post-mortem tissue from individuals with FXS and in the Fmr1 KO mouse have shown that functional deletion of FMRP leads to increased density of filopodia-like and immature dendritic spines (Irwin et al, 2001, 2002), suggesting an important role of FMRP for dendritic spine development and maintenance. The analyzed parameters and the described dendritic spine abnormalities vary between different reports. They include in vivo and in vitro studies, as well as analyses of dendritic protrusion and filopodia density, dendritic spine classification, and dendritic arborization (see, eg, McKinney et al, 2005; de Vrij et al, 2008; Gross et al, 2010; Grossman et al, 2010). Some findings are contradictory; for example, several studies have reported significantly increased total dendritic spine density in cultured hippocampal Fmr1 KO neurons as well as in cortex and olfactorial bulb in vivo (Hayashi et al, 2007; Gross et al, 2010; Liu et al, 2011; Scotto-Lomassese et al, 2011), whereas other studies have detected increased numbers of filopodia, but no significant difference in total spine density in the cortex in vivo and in cultured hippocampal neurons (Irwin et al, 2002; de Vrij et al, 2008; Bilousova et al, 2009). Different experimental settings, such as age of the mice or neuronal cultures, might explain variations in the observed phenotypes. An earlier study, for example, has shown that the dendritic spine phenotype in Fmr1 KO mice is age dependent (Nimchinsky et al, 2001). Although the exact parameters analyzed vary from study to study, abnormal dendritic spine morphology in Fmr1 KO mice is a robust phenotype and was observed in many different laboratories (for a recent review of the dendritic spine phenotype, see Portera-Cailliau, 2011).
Several studies in Fmr1 KO mice and dfmr1 Drosophila mutants have demonstrated that FMRP is important for the development and activity-dependent plasticity of neuronal connections. These reports have provided considerable insight into the mechanism that might underlie abnormal synapse development and dendritic spine morphology in FXS. In Drosophila, FMRP is essential for synapse formation and experience-dependent axonal pruning during development (Gatto and Broadie, 2008; Tessier and Broadie, 2008). Studies in Fmr1 KO mice have further revealed that FMRP regulates protein synthesis-dependent axon pruning, dendritic spine elimination, and actin-dependent stabilization of spines. In Fmr1 KO mice, disruption of this regulation leads to abnormal rates of dendritic spine turnover, delayed stabilization of dendritic spines during development, and absence of experience-induced dendritic spine modulation (Pfeiffer and Huber, 2007; Li et al, 2009; Chen et al, 2010; Cruz-Martin et al, 2010; Pan et al, 2010; Pfeiffer et al, 2010; Scotto-Lomassese et al, 2011).
The aberrant spine morphology in FXS might be due to loss of FMRP-dependent translational control of proteins necessary for synapse development or dendritic spine stability. One important group of FMRP-regulated proteins comprises cytoskeletal or scaffold proteins needed for synapse development and/or dendritic spine stability, such as MAP1B, PSD-95, or Shank (reviewed in Bassell and Warren, 2008). Dysregulation of these proteins in the absence of FMRP might contribute to the abnormal dendritic spine morphology in FXS. More recent studies reveal that the function of FMRP for microRNA-mediated translational control of specific targets such as NR2A and PSD95 is important for dendritic spine morphology (Edbauer et al, 2010; Muddashetty et al, 2011).
Furthermore, FMRP was proposed to regulate actin filament reorganization and stabilization mediated by the Rac/p21-activated kinase (PAK) signaling pathway (Chen et al, 2010). Interestingly, Chen et al (2010) also demonstrated that synaptic activity failed to induce the Rac/PAK pathway in Fmr1 KO mice. This suggests that the absence of experience-dependent dynamic changes of spine morphology in FXS might be because of abnormal neuronal signal transduction regulating the actin cytoskeleton. Of note, a dominant-negative PAK transgene rescued increased dendritic spine density in Fmr1 KO mice (Hayashi et al, 2007). However, it is not known whether the underlying mechanisms might involve loss of FMRP-mediated translational control of any Rac/PAK signaling components.
Most of the work analyzing the molecular mechanisms leading to abnormal spine morphology have been done in vitro in cultured neurons or in vivo in fixed tissue, which possibly limits their value. Only recently, studies have begun to analyze the function of FMRP for dendritic spine morphology in vivo in living mice (Cruz-Martin et al, 2010; Pan et al, 2010). These analyses revealed that dendritic spines are more transient and show delayed stabilization in Fmr1 KO mice (reviewed in Portera-Cailliau, 2011). In the future, more studies are needed to test whether the mechanisms observed in vitro can be recapitulated in vivo in living animals. In addition, the identification of specific FMRP target mRNAs important for regulating dendritic spine morphology will provide further insight into the causes of aberrant dendritic spine development and dynamics in FXS.
Although the underlying molecular mechanisms are not fully understood yet, the dendritic spine phenotype in Fmr1 KO mouse models has been proven to be an important readout to evaluate novel therapeutic strategies in FXS (Dolen et al, 2007; de Vrij et al, 2008; Bilousova et al, 2009; Gross et al, 2010; Liu et al, 2011).
Altered Neuronal Network Formation in FXS
Abnormal synapse development and morphology is expected to compromise synaptic function and might thus contribute to the observed alterations in neuronal circuit formation in FXS animal models. Gatto and Broadie (2009) reported abnormalities in circadian clock circuits in dfmr1 mutant Drosophila, which were due to aberrant synaptic architecture and could be rescued by temporally controlled overexpression of dFMRP. In Fmr1 KO mice, dysregulated neuronal connectivity in the barrel cortex (Bureau et al, 2008) leads to defective glutamatergic synapse maturation, delayed and aberrant formation of sensory maps, and altered synaptic plasticity during the critical period (Harlow et al, 2010). In general, loss of FMRP seems to lead to altered network synchrony and hyperexcitable neuronal networks (Chuang et al, 2005; Gibson et al, 2008).
The underlying mechanisms of network synchrony abnormalities are only poorly understood so far. Evidence from FXS Drosophila and mouse models suggests that calcium signaling is disturbed in the absence of FMRP, which could partially account for defects in neuronal network formation (Meredith et al, 2007; Tessier and Broadie, 2008). Furthermore, a recent report proposed a role for FMRP in retinoic acid-mediated homeostatic plasticity, an important signaling pathway regulating network activity (Soden and Chen, 2010). This suggests that dysregulated neuronal signal transduction contributes to altered network activity in FXS. Dysregulation of several FMRP targets might also contribute to aberrant network plasticity in FXS. Arc/Arg3.1, which was suggested to be an FMRP target (Zalfa et al, 2003; Krueger et al, 2011), was shown to be involved in neuronal network homeostasis (Shepherd et al, 2006; Peebles et al, 2010; Béïque et al, 2011). Moreover, lately, a few studies have reported that FMRP regulates potassium channels, which might also contribute to abnormal network activity in FXS. Expression and/or function of three different potassium channels have been suggested to be altered in the absence of FMRP: the sodium-activated potassium channel Slack (Brown et al, 2010), and the voltage-gated potassium channels Kv3.1b (Strumbos et al, 2010) and Kv4.2 (Gross et al, 2011).
Taken together, several studies have demonstrated that absence of FMRP leads to aberrant neuronal network homeostasis in animal models. Additional studies are needed to investigate if and how FMRP directly influences neuronal network synchrony, for example, by regulation of the function or protein expression of specific targets.
Altered Synaptic Plasticity in Fmr1 KO Mice
The dysfunctions in neuronal network formation might contribute to dysregulated synaptic plasticity observed in FXS. The most prominent and best-studied example is hippocampal metabotropic glutamate receptor 1/5 (mGlu1/5)-dependent synaptic long-term depression (LTD), which is exaggerated and, in contrast to wild type, protein synthesis independent in Fmr1 KO hippocampus (Huber et al, 2002; Nosyreva and Huber, 2006; Hou et al, 2006; further discussed below). Several studies have also shown impaired long-term potentiation (LTP) in different cortical areas of Fmr1 KO mice (Li et al, 2002; Zhao et al, 2005; Desai et al, 2006). In contrast, LTP appeared to be normal in the Fmr1 KO hippocampus (Paradee et al, 1999; Li et al, 2002; Larson et al, 2005). However, a defect in hippocampal LTP could be detected at lower levels of stimulation (Lauterborn et al, 2007), which was in line with another study showing that spike-timing-dependent LTP in the Fmr1 KO cortex was normal after a strong stimulus, but impaired when using a threshold induction paradigm (Meredith et al, 2007). Together, these findings suggest that in the absence of FMRP, neurons are capable of developing LTP, but require a stronger stimulus than wild-type neurons. In addition, more recently, specific mGlu1/5-dependent forms of LTP and LTP priming were shown to be altered in the Fmr1 KO hippocampus (Shang et al, 2009; Auerbach and Bear, 2010; further discussed below). Apart from hippocampus and cortex, impaired long-term synaptic plasticity has also been shown in the amygdala (reviewed in Suvrathan and Chattarji, 2011) and the cerebellum of Fmr1 KO mice (reviewed in Huber, 2006).
In summary, loss of FMRP leads to impairments in several forms of long-term synaptic plasticity in different brain areas in Fmr1 KO mice. Future studies analyzing, for example, which phases of LTP are affected in the Fmr1 KO brain might identify the signal transduction pathways involved in defective synaptic plasticity in Fmr1 KO mice, and may help to develop therapeutic strategies to treat cognitive impairments in patients with FXS.
The Behavioral Phenotype of Fmr1 Mouse Models
FXS is a complex disorder and patients suffer from a variety of neurological problems, such as intellectual disabilities, epilepsy, and autism. The behavioral characterization of the FXS mouse model has covered several aspects of this disease pattern including: (1) cognitive performance, (2) seizure susceptibility and general neuronal activity, (3) sensitivity to sensory stimuli, and (4) socioemotional behavior. In general, the evaluation of behavioral phenotypes in Fmr1 KO mice has been challenging because the observed defects are rather subtle and seem to strongly depend on genetic background, age, and experimental settings (reviewed, eg, in Bernardet and Crusio, 2006; Bhogal and Jongens, 2010). Initial studies using ‘classic’ hippocampus-dependent learning tasks, such as Morris Water Maze and fear conditioning, revealed no or very subtle defects in the Fmr1 KO mice (The Dutch-Belgian Fragile et al, 1994; Paradee et al, 1999; Van Dam et al, 2000). Interestingly, a recent report has shown that prefrontal cortex-dependent forms of learning are altered in Fmr1 KO mice (Krueger et al, 2011), and patients with FXS are specifically impaired in several prefrontal cortex-dependent cognitive skills (Hoeft et al, 2007; Reiss and Hall, 2007; Mercaldo et al, 2009). This suggests that behavioral paradigms testing prefrontal cortical function in Fmr1 KO mice might be a better tool to assess therapeutic strategies for their potential to rescue cognitive impairment in FXS (Krueger et al, 2011).
The most promising behavioral assays to evaluate new rescue strategies in FXS mouse models are those that are targeted on neuronal hyperexcitability and have correlates in human patients, such as epilepsy and hyperactivity. One of the most robust assays tests the susceptibility to audiogenic seizures. Higher susceptibility to audiogenic seizures is reliably observed in Fmr1 KO mice of various genetic backgrounds and has become very useful to evaluate potential therapeutic strategies (Musumeci et al, 2000; Yan et al, 2004, 2005; Dolen et al, 2007; Min et al, 2009). Recently, a kindling paradigm similarly revealed differences in seizure susceptibility in wild-type and Fmr1 KO mice (Qiu et al, 2009). If reproducible, this seizure paradigm might become an additional tool to assess hyperexcitability in the future. Apart from seizure activity, altered open field activity in mice was reproduced by several laboratories. Although this phenotype is less robust and seems to vary with age and experimental settings (Bernardet and Crusio, 2006), it was used successfully to test potential therapeutic targets for FXS (Spencer et al, 2005; Yan et al, 2005; Yuskaitis et al, 2010). Additionally, behaviors that phenocopy the hypersensitivity to sensory stimuli observed in FXS patients, such as altered auditory startle responses and impaired prepulse inhibition, have been reproduced in several laboratories and used to show the efficacy of drug treatments (reviewed in Bernardet and Crusio, 2006; Brodkin, 2008). Of interest, Fmr1 KO mice display autistic-like behaviors and have been suggested as valuable mouse model for autism (Bernardet and Crusio, 2006; discussed in Box 1).
In the future, behavioral assays in Fmr1 KO mice will have to be better standardized for experimental paradigms, age, and genetic background to provide a valuable and reproducible tool for the evaluation of novel therapeutic strategies in FXS.
The characterization of FXS animal models suggests that FMRP regulates diverse neuronal functions and mechanisms. As detailed below, many of the described neuronal defects can be attributed to the function of FMRP as a major regulator of neuronal signal transduction controlling protein synthesis at the synapse. Therefore, dysregulated signaling pathways in the absence of FMRP are promising targets for therapeutic interventions in patients with FXS.
THE mGluR THEORY: THERAPEUTIC VALUE AND LIMITATIONS
Increased and Protein Synthesis-Independent mGlu1/5 LTD in FXS
In 2002, a seminal study by Huber et al (2002)reported that Glu1/5-mediated LTD, a specific protein synthesis-dependent form of synaptic plasticity in the hippocampus, is elevated in Fmr1 KO mice. This discovery motivated the formulation of the mGluR theory of FXS, which posits that dysregulated mGlu1/5-mediated protein synthesis-dependent forms of synaptic plasticity contribute to the pathology of FXS (Bear et al, 2004). Hence, Bear et al (2004) proposed that mGlu1/5 signaling might be a promising therapeutic target for FXS. Follow-up studies further demonstrated that, in contrast to wild-type, mGlu1/5 LTD is protein synthesis-independent in Fmr1 KO mice (Nosyreva and Huber, 2006; Hou et al, 2006), thus corroborating the assumption that a major regulator of neuronal protein synthesis is lost in FXS.
A crucial molecular mechanism underlying induction of mGlu1/5-mediated LTD is the protein synthesis-dependent increase of GluA endocytosis (for a detailed review, see Gladding et al, 2009). This mechanism appears to be compromised in FXS, because reduced levels of FMRP were shown to lead to increased mGlu5-mediated GluA endocytosis in rat hippocampal neurons (Nakamoto et al, 2007). The mGlu1/5-dependent translational induction of two FMRP targets, Arc/Arg3.1 and MAP1B, which were suggested to be involved in activity-regulated GluA endocytosis in hippocampal neurons (Davidkova and Carroll, 2007; Waung et al, 2008), is absent in Fmr1 KO mice (Hou et al, 2006; Park et al, 2008). Waung and Huber (2009) thus hypothesized that in the absence of the translational repressor FMRP, several ‘LTD-proteins’ are already present in excess at the synapse before LTD-inducing stimuli and could contribute to the exaggerated LTD phenotype. This hypothesis is in line with previous studies suggesting increased basal protein expression of MAP1B in the hippocampus and Arc/Arg3.1 in total brain fractions from Fmr1 KO mice (Zalfa et al, 2003; Lu et al, 2004). However, in contrast to Zalfa et al (2003), another study reported reduced Arc/Arg3.1 protein levels in the orbital frontal cortex and medial prefrontal cortex of FXS mice (Krueger et al, 2011). Future experiments are needed to analyze whether FMRP regulates specific target mRNAs differently depending on the brain region.
Taken together, these studies suggest that the role of FMRP in regulating mGlu1/5-induced protein synthesis might underlie the exaggerated LTD phenotype in FXS mice. In the future, further identification and analysis of FMRP target mRNAs that have a function for mGlu1/5 LTD will help to fully understand the mechanisms leading to dysregulated LTD in FXS.
Other mGlu1/5-Dependent Mechanisms in FXS
In support of the mGluR theory, several studies have demonstrated that mGlu1/5-mediated neuronal and synaptic protein synthesis are elevated and stimulus-insensitive in Fmr1 KO cortex and hippocampus (Dolen et al, 2007; Muddashetty et al, 2007; Gross et al, 2010; Osterweil et al, 2010). Subsequent work also suggested that apart from hippocampal LTD, additional mGlu1/5-dependent neuronal mechanisms are dysregulated in the FXS brain. In the hippocampus, deletion of FMRP was shown to lead to prolonged epileptiform discharges caused by enhanced sensitivity of mGlu1/5 to synaptically released glutamate (Chuang et al, 2005). Loss of FMRP also leads to changes in mGlu1/5-mediated endocannabinoid (eCB) synthesis and release in the hippocampus (Zhang and Alger, 2010). Several other brain regions display altered mGlu1/5-dependent signaling. MGlu1/5 signaling was shown to be dysregulated in the anterior cingulate cortex of Fmr1 KO mice (Wang et al, 2008a, 2009). Moreover, Harlow et al (2010) demonstrated impaired synaptic plasticity in the barrel cortex in Fmr1 KO mice during the critical period, a process that was shown to involve FMRP and mGlu1/5 activity (Todd et al, 2003). Another study reported aberrant mGlu1/5-dependent neuronal plasticity in the amygdala of Fmr1 KO mice (Suvrathan et al, 2010). Furthermore, a specific, mGlu1/5- and protein synthesis-dependent form of hippocampal LTP was shown to be dysregulated in Fmr1 KO mice (Shang et al, 2009), and a recent study demonstrated that mGlu1/5-mediated priming of hippocampal LTP is, in contrast to wild-type, protein synthesis-independent in Fmr1 KO mice (Auerbach and Bear, 2010). Apart from LTD in the hippocampus, mGlu1/5-dependent LTD was also shown to be exaggerated in the cerebellum in FXS mice (Koekkoek et al, 2005; Huber, 2006). The defects in mGlu1/5-dependent pathways in these different brain areas support speculations that dysregulated mGlu1/5 signaling in patients with FXS might not only underlie cognitive impairments, but also epilepsy (epileptiform discharges in the hippocampus), hypersensitivity to sensory stimuli (barrel cortex), anxiety (amygdala), and motor learning defects (cerebellum). Besides, Price et al (2007) showed that mGlu1/5-dependent nociception in the spinal cord of Fmr1 KO mice is altered and proposed that this may contribute to self-injurious behavior and altered perception of sensory stimuli and pain in patients. The correlation of phenotypes in animal models of FXS with those in humans is in most parts still hypothetical, but nevertheless these studies support the hypothesis that therapies reducing mGlu1/5 signaling might ameliorate several different symptoms in patients.
In line with these findings of dysregulated mGlu1/5 signaling in numerous paradigms, several rescue studies in mouse (Yan et al, 2005; Dolen et al, 2007) and Drosophila (McBride et al, 2005) models provided compelling support of the mGluR theory. These studies showed that both pharmacologic and genetic reduction of mGlu5 signaling could ameliorate or reverse FXS-associated phenotypes in vivo and in vitro (reviewed in Levenga et al, 2010).
The underlying mechanisms of increased and stimulus-insensitive mGlu1/5 signaling are still being explored, and findings are partially controversial. In general, so far, there is no compelling evidence that FMRP directly regulates mGlu1/5 receptor expression or function. A study by Giuffrida et al (2005) showed that in Fmr1 KO mice, less mGlu1/5 receptors are associated with the scaffolding protein Homer. Homer links mGlu1/5 to the phosphoinositide-3 kinase (PI3K) enhancer PIKE and the catalytic PI3K subunit p110. This is an alternative pathway to activate PI3K signaling downstream of mGlu1/5, apart from activation via heterotrimeric G-proteins (Kurosu et al, 1997; Rong et al, 2003) (also see Figure 1). Interestingly, Ronesi and Huber (2008) reported that, in contrast to wild-type, neither mGlu5–Homer interactions nor mGlu1/5-mediated activation of PI3K signaling are necessary to elicit LTD in Fmr1 KO mice. Although induction of PI3K signaling was impaired in Fmr1 KO hippocampus, no elevated basal PI3K downstream signaling could be detected. This finding is in line with a report from Osterweil et al (2010), but in contrast to two other recent studies showing excess basal PI3K activity and downstream signaling (Gross et al, 2010; Sharma et al, 2010). These apparent contradictions might be because of differences in the examined tissue, but further analyses are needed to resolve these discrepancies and understand the molecular mechanisms of impaired mGlu1/5–Homer–PI3K signaling. In summary, the above discussed studies suggest that mGlu1/5 might be uncoupled from downstream signaling in FXS, which could, for example, be explainable by a role of FMRP for regulating signal transduction.

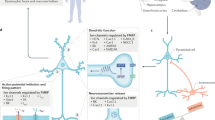
Dysregulated signal transduction as therapeutic target in FXS. Shown are signaling pathways and molecules that have proven to be promising targets for therapeutic treatment based on preliminary clinical trials and/or studies in FXS animal models. Targets can be divided into three major groups: (1) membrane receptors such as GABA, mGlu1/5, and dopamine D1/5 receptors and their regulator RGS4; (2) the central intracellular signaling molecules ERK1/2 and PI3K; and (3) downstream targets such as MMP-9, mTOR, GSK3β, GRK2, and PAK. Although genetic and pharmacologic studies suggest that these proteins might be promising therapeutic targets, the underlying mechanisms are mostly elusive, with a few exceptions: FMRP was shown to directly regulate PI3K and PIKE mRNA translation, protein expression, and enzymatic activity (indicated by red outlines) (Gross et al, 2010; Sharma et al, 2010). Furthermore, FMRP associates with PAK and GRK2 protein, but the functional consequences of these interactions are unknown (Hayashi et al, 2007; Wang et al, 2008b). Some potential therapeutic targets show dysregulated expression (MMP-9, GABA receptors), subcellular localization (PAK and GRK2), and/or phosphorylation (ERK1/2, GSK3β, PAK, GRK2, mTOR) in the absence of FMRP (highlighted with yellow asterisks), but the detailed mechanisms are unknown and future studies will have to show whether these are the direct effects of loss of FMRP or are caused by dysregulated signaling through other pathways. Yellow triangles mark mGlu receptors and the regulator of G-protein signaling RGS4, which were shown to be promising therapeutic targets, but do not seem to be directly altered by loss of FMRP. Black arrows indicate how upstream membrane receptors activate and/or regulate some of the downstream targets, illustrating crosstalk between pathways and putative shared dysregulated downstream mechanisms: MMP-9 was suggested to be regulated by mGlu1/5 signaling (Bilousova et al, 2009) and by GABAergic signaling via an ERK-dependent mechanism (Miao et al, 2010). ERK1/2 and PI3K are regulated by mGlu1/5 (Banko et al, 2006) and by TrkB signaling (Yoshii and Constantine-Paton, 2010). ERK1/2 was shown to signal downstream of D1/5 (Beaulieu and Gainetdinov, 2011). A study suggests that PI3K signaling might affect GRK2 activity (Banday et al, 2007), which might contribute to the crosstalk between mGlu1/5 and D1/5 (Deng et al, 2010). GRK2 was suggested to be involved in dysregulated signaling through D1 receptors in Fmr1 KO mice (Wang et al, 2008b). PI3K can regulate PAK activity via Rac and ERK1/2 itself was shown to be regulated by Rac/PAK signaling (Lim et al, 1996; Welch et al, 2003). GSK3β activity is affected by both ERK1/2 and PI3K, downstream of mGlu1/5 and D1/5 (Lebel et al, 2009; Min et al, 2009).
Taken together, although the underlying molecular mechanisms are still under investigation, it has become evident from several studies that FMRP plays an essential role for stimulus-induced mGlu1/5 signaling. The successful validation of the mGluR theory in different animal models and paradigms further substantiates the value of mGlu1/5 as a therapeutic target in FXS, and has led to the development of mGlu5-specific antagonists and the initiation of clinical trials targeting mGlu5 signaling in FXS.
Altered Signaling of Other Membrane Receptors
Dysregulated signaling through mGlu1/5 was one of the first molecular mechanisms shown to contribute to the pathology of FXS. Later on, several other neurotransmitter receptor-mediated signal transduction pathways regulating neuronal protein synthesis have been demonstrated to be affected in FXS. Aberrant signaling in the absence of FMRP was shown to occur through (1) other Gq protein-coupled receptors such as muscarinic acetylcholine receptors (Volk et al, 2007, 2) the dopamine D1/5 receptors (Wang et al, 2008b, 2010a), and (3) tyrosine kinase receptor B (TrkB) (Osterweil et al, 2010). Of note, these signaling pathways employ similar molecular mechanisms like mGlu1/5-induced LTD. Activation of signaling through both Gq-coupled receptors and dopamine D1 receptors induces protein synthesis-dependent GluA receptor endocytosis (Volk et al, 2007; Wang et al, 2010a). Likewise, TrkB signaling has also been shown to stimulate protein synthesis and regulate GluA receptor surface expression (Caldeira et al, 2007; Minichiello, 2009). Similar to mGlu1/5 signaling in FXS, altered signal transduction mediated by these receptors is thus believed to be caused by the lack of activity-regulated translational control in the absence of FMRP.
Several studies suggested modulation of GABAergic inhibitory signaling as an alternative or additional therapeutic strategy in FXS: Fmr1 KO mice show impaired GABAergic inhibition in several different brain areas (Centonze et al, 2007; Curia et al, 2009), which is in line with the notion that the absence of FMRP converts neurons into a ‘hyperexcitable’ state. Impaired GABAergic signaling in FXS might partially be caused by decreased expression of GABA receptors in these brain areas (El Idrissi et al, 2005; D'Hulst et al, 2006; Adusei et al, 2010). On the other hand, it could be a secondary effect of dysregulated mGlu1/5-dependent signaling, because these two receptors were shown to crosstalk in several different brain areas (Hirono et al, 2001; Deng et al, 2010; Kolaj and Renaud, 2010). The exact mechanisms of reduced GABA-mediated signaling in FXS are currently unclear; nevertheless, results from animal studies and clinical trials suggest that GABA agonists are suitable to treat FXS-associated symptoms (Chang et al, 2008; Heulens et al, 2010; Olmos-Serrano et al, 2010).
Overall, these findings have two important implications: first, they have revealed additional promising therapeutic targets in FXS, and second, they provide important mechanistic insights suggesting that FMRP might regulate a downstream mechanism shared by several signal transduction pathways involved in stimulus-dependent protein synthesis and neuronal plasticity.
FXS EMERGES AS A DISEASE OF DYSREGULATED SIGNALING
In light of these and other recent findings, FXS emerges from being a disease of dysregulated mGlu1/5 signaling to a disease of dysregulated general neuronal signaling upstream of protein synthesis (summarized in Figure 1). In support of this hypothesis, FMRP was shown to regulate general stimulus-induced neuronal and synaptic protein synthesis (Weiler et al, 2004; Qin et al, 2005; Dolen et al, 2007; Muddashetty et al, 2007; Gross et al, 2010; Osterweil et al, 2010). A study by Napoli et al (2008) suggested that FMRP directly regulates translation initiation by interaction with CYFIP1, which binds to and inhibits the translation initiation factor eIF4E. In addition, one could speculate that specific target mRNAs of FMRP might encode translation factors or intracellular signal transducers that are important for activity-dependent protein synthesis. As discussed below, recent studies support the hypothesis that FMRP might regulate key signaling molecules involved in translation initiation.
Targeting Downstream Signaling Molecules in FXS
In support of the hypothesis that FMRP has a crucial function for neuronal signal transduction, the genetic or pharmacologic inhibition of several molecules downstream of neurotransmitter-induced signaling have been shown to reverse relevant phenotypes in FXS animal models (Figure 1). Those proteins included not only glycogen-synthase kinase 3β (GSK3β), metallomatrix protease 9 (MMP-9), PAK, and regulator of G-protein signaling 4 (RGS4), but also key cellular signal transducers such as extracellular signal-regulated kinase 1/2 (ERK1/2) and PI3K.
Fmr1 KO mice display elevated and dysregulated GSK3β activity, a downstream signaling target of mGlu1/5 (Min et al, 2009; Yuskaitis et al, 2010). Several studies demonstrated that lithium, which among other things antagonizes GSK3α and GSK3β activity (Berridge et al, 1989; De Sarno et al, 2002), could rescue dysregulated GSK3β activation, aberrant dendritic spine morphology, and several behavioral phenotypes in Fmr1 KO mice (Liu et al, 2011; Mines et al, 2010; Yuskaitis et al, 2010), as well as cognitive impairments in a Drosophila model of FXS (Choi et al, 2010). Pharmacologic studies in Fmr1 KO mice also suggested that the MMP-9 inhibitor minocycline might be a useful drug to ameliorate FXS symptoms (Bilousova et al, 2009). MMP-9 protein levels were shown to be increased by mGlu1/5 stimulation in wild-type mice and under basal conditions in Fmr1 KO mice (Bilousova et al, 2009). Future studies will have to show whether MMP-9 is a direct target of FMRP or whether the dysregulated expression of MMP-9, and thus the therapeutic benefits of minocycline, are a treatable indirect effect of dysregulated signaling downstream of mGlu1/5 receptors in FXS. MMP-9 was also shown to be regulated by GABA signaling via an ERK1/2-dependent mechanism (Tyagi et al, 2009). Thus, MMP-9 antagonists might improve dysregulated signaling through several pathways. Both lithium and minocycline have already been tested in clinical studies in patients with FXS (Berry-Kravis et al, 2008a; Paribello et al, 2010, see below).
Genetic rescue studies in Fmr1 KO mice suggest that PAK and RGS4 might be putative therapeutic targets in FXS, but need to be confirmed in independent studies. Postnatal expression of a dominant-negative form of PAK reversed increased dendritic spine density and several behavioral phenotypes in Fmr1 KO mice (Hayashi et al, 2007), motivating the development of PAK-specific inhibitors (www.afraxis.com). Furthermore, deletion of RGS4, a protein involved in downstream signaling of G-coupled receptors, such as GABA and mGlu receptors, in Fmr1 KO mice could rescue altered social behavior, increased body weight, and aberrant synaptic translation (Pacey et al, 2011). However, other Fmr1 KO phenotypes, such as hyperactivity and macroorchidism, were not affected by knockout of RGS4, suggesting that these might be regulated by different or additional molecular mechanisms.
In summary, these studies show that the loss of FMRP leads to dysregulation of multiple signaling pathways, and extend the list of potentially valuable targets for therapeutic treatments in patients with FXS. These findings also suggest that FMRP might control shared signaling molecules that are key players to regulate neurotransmitter-mediated signaling and protein synthesis. The two major pathways regulating neurotransmitter-induced protein synthesis in neurons are the ERK1/2 and the PI3K/mTOR (mammalian target of rapamycin) pathways (Banko et al, 2006; Nagai et al, 2007; Schicknick et al, 2008; Santos et al, 2010; Zhou et al, 2010). Both pathways also play crucial roles for intracellular signaling, and many of the above discussed dysregulated neurotransmitter-dependent signaling mechanisms in FXS are regulated or mediated via ERK1/2 and/or PI3K signaling (Figure 1). Therefore, antagonizing central downstream signaling molecules like ERK1/2 or PI3K might correct a broad variety of different FXS-associated symptoms caused by any of these signaling defects. ERK1/2 or PI3K antagonists could offer an alternative approach to therapeutic strategies targeting an upstream neurotransmitter receptor or downstream signaling molecules that are restricted to specific pathways.
PI3K and ERK Signaling as Therapeutic Targets
In support of the hypothesis that ERK1/2 and PI3K might be promising therapeutic targets in FXS, several recent studies suggest a function for FMRP in controlling the activity of ERK1/2 and PI3K/mTOR signaling pathways. Currently, the findings regarding ERK1/2 and PI3K/mTOR signaling in FXS are controversial. Several studies report deficient mGlu1/5-mediated ERK1/2 activation (Hou et al, 2006; Kim et al, 2008; Weng et al, 2008; Osterweil et al, 2010), whereas others have shown that the PI3K/mTOR signaling response to mGlu1/5 stimulation is lost (Ronesi and Huber, 2008; Gross et al, 2010; Sharma et al, 2010). Most of these studies analyze phosphorylation of downstream targets to evaluate basal and stimulus-induced activity of PI3K and ERK1/2. In general, these results have to be evaluated carefully, as ERK1/2 and PI3K/mTOR signaling pathways are not separate entities, but rather exist in a signaling network, implying that dysregulation of one of these pathways in FXS might result in aberrant signaling through the other pathway. Of note, a direct comparison of basal enzymatic activity of ERK1/2 and PI3K showed excess PI3K activity, but normal ERK1/2 activity in Fmr1 KO mice (Gross et al, 2010). As a possible mechanism underlying excess PI3K activity, FMRP was shown to regulate protein expression of two PI3K activity-modulating enzymes, the catalytic PI3K subunit p110β and the PI3K enhancer PIKE (Gross et al, 2010; Sharma et al, 2010), whereas the mechanisms underlying dysregulated ERK1/2 signaling remain elusive. However, both ERK1/2 and PI3K antagonists were shown to rescue phenotypes in FXS mice: intraperitoneal injection with an ERK1/2 inhibitor prevented audiogenic seizures in Fmr1 KO mice (Osterweil et al, 2010), and PI3K inhibitors reduced excess dendritic spine density and aberrant GluA receptor endocytosis in Fmr1 KO hippocampal neurons (Gross et al, 2010). The results analyzing the effect of ERK1/2 and PI3K/mTOR inhibitors on dysregulated protein synthesis in FXS were contradictory in these studies, and future, independent studies are needed for validation and clarification. Of interest for their evaluation as therapeutic treatment, a PI3K inhibitor, but not an ERK1/2 inhibitor, could restore mGlu1/5-induced increase in synaptic protein synthesis (Gross et al, 2010). Likewise, antagonizing PI3K activity restored mGlu1/5-mediated mTOR activation (Sharma et al, 2010). Responsiveness of neurons to external stimuli is an important prerequisite for synaptic plasticity, and a successful therapeutic strategy for FXS will have to not only reduce excess basal signaling, but also restore neuronal response to activity-inducing stimuli. These initial results are therefore promising to further explore PI3K inhibitors as therapeutic targets.
A safety concern for inhibition of PI3K and ERK1/2 in humans is their essential role for cell proliferation and apoptosis. The broad-spectrum PI3K or ERK1/2 antagonists, as used in the discussed studies, could potentially induce severe side effects. Mutations causing aberrant signaling through PI3K/mTOR and/or ERK1/2 have been shown to lead to several different forms of cancer. However, so far, unchanged (Sund et al, 2009) or rather decreased (Schultz-Pedersen et al, 2001) cancer rates have been reported in patients with FXS. This suggests that in FXS, a very specific function of ERK1/2 and/or PI3K/mTOR signaling is dysregulated, which might be different from cell proliferation and apoptosis. The use of specific antagonists targeting these functions could limit or eliminate side effects and might, thus, be suitable for pharmacologic treatment in human patients with FXS. In the case of PI3K signaling, promising candidates for specific inhibition are the two FMRP target mRNAs: PIKE and p110β. Interestingly, p110β-specific antagonists are already being developed and tested for the treatment of certain forms of cancer (Blajecka et al, 2011). Future studies are needed to test whether ERK1/2 pathway components might be promising therapeutic targets in FXS as well, and whether genetic or pharmacologic reduction of p110β, PIKE, or ERK1/2 can rescue FXS-specific phenotypes in animal models.
Although initial studies showing rescue of FXS-associated phenotypes with ERK1/2 and PI3K antagonists are promising, they will have to be confirmed and extended to more specific antagonists before a potential application in clinical trials with human patients. Importantly, future studies will have to show whether PI3K-specific inhibition can rescue behavioral phenotypes in Fmr1 KO mice. Furthermore, additional mechanistic studies are needed to solve the current controversies about basal and activity-induced ERK1/2- and PI3K-mediated signaling in the absence of FMRP.
Taken together, these genetic and pharmacologic rescue studies antagonizing different signal transduction molecules in FXS animal models have provided important rationale for novel pharmacologic interventions in FXS. Most of these novel therapeutic strategies will have to be further tested in preclinical studies before clinical tests in humans, but they suggest that in the future, they might become alternative or additional disease-targeted treatments for FXS patients.
THERAPEUTIC INTERVENTION IN FXS
Preclinical data with targeted agents acting on the mGlu1/5, GABA, or other neuronal pathways found to be dysregulated in FXS model systems have been very promising. Although there is much less information on the impact of treatment of humans with FXS with these compounds, early-phase clinical trials have been initiated for some of these targeted treatments. This early translational work is described below, in sections corresponding to mechanistic categories of the proposed targeted treatment.
Agents Reducing Overactive Signaling Between Receptors and the Dendritic Translational Machinery
Agents with extracellular targets
Several mGlu5 negative allosteric modulators (NAMs) are currently being developed for treatment of FXS and are in clinical trials. Fenobam, the first mGlu5 NAM used in FXS, was administered in a single oral dose to a cohort of 12 adult males and females with FXS (Berry-Kravis et al, 2009). An improvement in the general anxiety level of many of the participants was observed after the dose, although given the trial was open label, this could have been a placebo effect. A significant improvement in PPI was also seen, which would be far less likely because of the placebo effect. There were no safety concerns. Concurrently, a phase II double-blind placebo-controlled, crossover design trial of AFQ056 (Novartis), with treatment of 30 adult males with FXS for 28 days each with AFQ and placebo, was conducted in Europe (Jacquemont et al, 2011). The results of this trial suggested improvement in maladaptive behavior on the Aberrant Behavior Checklist-Community Edition (ABC-C), Clinician Global Impression of Improvement (CGI-I) scale, and the Repetitive Behavior Scale in the subgroup of males with full methylation of FMR1. This outcome was adequate to support ongoing development of AFQ056 for treatment of FXS, with a larger multinational double-blind placebo-controlled 3-month trial evaluating the effects of multiple doses of AFQ056 started in the Fall of 2010 (clinicaltrials.gov). Phase I trials of STX107 (Seaside Therapeutics) have been completed in healthy individuals and early treatment trials in FXS are expected in 2011 (clinicaltrials.gov). A double-blind placebo-controlled dose-finding phase II trial of RO4917523 (Roche) in adult males and females with FXS is underway and should be completed by mid-2011 (clinicaltrials.gov). No serious safety concerns have yet emerged in any of these studies, although only a small population of individuals with FXS has thus far been exposed to mGlu5 NAMs. It has been proposed that mGlu1 (Koekkoek et al, 2005) and muscarinic acetylcholine receptor (Volk et al, 2007) NAMs might also be beneficial for individuals with FXS, but these have not been studied. Motor and cognitive side effects were observed in rats exposed to mGlu1 NAMs (Steckler et al, 2005; Kolasiewicz et al, 2009), raising concerns about the possible toxicity in human trials.
Intracellular agents
Although a number of intracellular treatment targets have been proposed, in most cases safe and available agents acting on these targets are not yet developed for use in humans. One exception is lithium, for which the preclinical findings in the dfmr mutant fly and Fmr1 KO mouse suggested promise of therapeutic benefit. The effects of short-term (2 months) treatment with lithium were systematically explored for a broad range of phenotypes including behavior, cognition, and biophysical measures in a pilot open-label trial in 15 subjects with FXS (Berry-Kravis et al, 2008a) in order to test the concept of inhibition of mGlu1/5-activated translational signaling pathways as a treatment strategy for FXS. Significant improvement in behavior was seen with lithium on the total ABC-C score and three of the ABC-C subscales, the Maladaptive Behavior subscore from the Vineland Adaptive Behavior Scale, a parent visual analog scale for target behaviors, the CGI, and in verbal memory on the RBANS List Learning task. In addition, ERK1/2 phosphorylation rates, shown to be reduced in lymphocytes from humans with FXS (Weng et al, 2008), were normalized during lithium treatment (Berry-Kravis et al, 2008a), suggesting that ERK1/2 activation rates could be a biomarker for measuring changes in signaling during treatment with agents targeted to receptor-activated translational regulatory pathways. The side effects were generally mild to moderate and included polydipsia, polyuria, and abnormal thyroid measurements in a few subjects. These data suggested that further studies with a placebo-controlled trial would be indicated; however, such studies have not yet been carried out, partly because of concerns about the chronic toxicity of lithium, but also related to hope that less toxic mechanism-based treatments will be available soon.
Inhibitors of Individual Proteins Excessively Produced or Activated in the Absence of FMRP
Minocycline has been used in clinical trials to inhibit MMP-9, which shows increased activity in the Fmr1 KO mouse (Bilousova et al, 2009). An open-label trial of minocycline in 20 participants with FXS aged ⩾12 years showed behavioral improvements on the ABC-C, VAS, and CGI (Paribello et al, 2010). There were no major side effects but two individuals had to stop treatment because of elevated antinuclear antibodies (ANAs), despite lack of signs of drug-induced lupus. These results have been the rationale for an in-progress double-blind placebo-controlled trial of minocycline in children and adolescents with FXS aged ⩾5 years. The high risk of irreversible yellow/brown discoloration of the permanent teeth in children aged <12 years exposed to minocycline, coupled with the idea that targeted treatments might work better if applied at younger ages, has made it crucial to understand the true margin of benefit for this treatment. The other side effects that can be seen at any age include GI symptoms such as vomiting and/or diarrhea (Utari et al, 2010), drug-induced lupus, and pseudotumor cerebri.
Agents Activating Surface AMPA Receptors and Activity
A double-blind placebo-controlled trial of effects of CX516 (Cortex Pharmaceuticals) on safety and cognitive and behavioral efficacy measures was carried out in a cohort of 49 individuals with FXS (Berry-Kravis et al, 2006). CX516 is a direct AMPA receptor positive modulator or ‘ampakine’ known to increase LTP and raise BDNF levels, thus potentially increasing surface expression of AMPA receptors (Jourdi et al, 2009). Conceptually, it was thought that CX516 would help compensate or correct the AMPA receptor deficit resulting from mGlu pathway overactivity. Realistically, CX516 is a very weak ampakine and thus no improvements were seen except in the group of patients co-treated with an antipsychotic (known to potentiate ampakine activity). This suggests that a more potent ampakine molecule might be helpful in FXS when these become available.
Agents Acting on Other Receptors/Proteins That Regulate Synaptic Activity
In addition to promising findings in FXS animal models with racemic baclofen treatment and evidence that the GABA system is downregulated in the Fmr1 KO mice, anecdotal clinical experience in humans with FXS suggested potential behavioral benefits from racemic baclofen, and led to a clinical trial of R-baclofen in humans with FXS. R-baclofen is the enantiomer of racemic baclofen with more potent GABAB agonist activity. This double-blind placebo-controlled crossover trial (Seaside Therapeutics) with 4-week periods of placebo and active R-baclofen treatment for each subject (N=63) did not meet its primary end point of improvement on the ABC Irritability subscale. However, as a phase II exploratory trial targeted to an underlying molecular mechanism rather than a specific behavior, the behavioral target was actually not known. Thus, defining a primary end point a priori was difficult, and in post hoc analyses the trial did identify a different area of dysfunction in FXS, which R-baclofen appears to target. There was significant improvement during R-baclofen treatment relative to placebo in global preference for treatment period, CGI, the ABC Social Withdrawal subscale, and Vineland Socialization Scale in the subgroup of participants with FXS and more severe impairment in social behaviors (Wang et al, 2010b). There were no significant safety issues, and many subjects are continuing treatment though an extension study to evaluate the long-term benefits and toxicity. The positive effects of R-baclofen on social behavior will require confirmation in a larger study, and further development of R-baclofen is underway with two phase III placebo-controlled clinical trials (clinicaltrials.gov).
Other agents acting at an array of receptors have undergone exploratory study in groups with FXS. A pilot open-label study of NMDA antagonist memantine in six participants with FXS showed modest improvement on a CGI in four out of six patients, but no improvement on behavioral rating scales, and two patients showed treatment-limiting irritability (Erickson et al, 2009). An open-label study of a sodium channel blocker and glutamate uptake activator riluzole in five patients with FXS showed overall behavioral improvement in only one subject, although ERK1/2 activation rates normalized and there was a suggestion of improvement in hyperactivity symptoms across the group (Erickson et al, 2011b). An anecdotal study of three adults with FXS treated with acamprosate, a drug approved for alcohol withdrawal that is a mixed NMDA agonist/antagonist, GABAA receptor activator, and possible inhibitor of group I mGlus, reported promising improvement in language and behavior in all patients (Erickson et al, 2010), but limiting gastrointestinal side effects in one patient. Although aripiprazole is a treatment directed primarily at behavior rather than molecular mechanism, it could theoretically be targeted to dopamine deficits thought to be present in FXS (Wang et al, 2008b), given its dopamine agonist activity at lower doses. Aripiprazole has shown good success when used empirically in FXS clinic populations (Berry-Kravis and Potanos, 2004; Hagerman et al, 2009), and has resulted in improvement in the ABC Irritability subscore, other ABC subscores, and other behavioral rating scales in a pilot open-label trial in 15 individuals with FXS (Erickson et al, 2011a). Plans are underway to initiate a double-blind placebo-controlled trial of aripiprazole in FXS.
TRIAL DESIGN AND HURDLES IDENTIFIED IN EARLY CLINICAL TRIALS IN HUMANS WITH FXS
Although many neuronal targets for treating the underlying disorder in FXS have emerged, and early translational work has begun, there are still many uncertainties about how to best demonstrate the treatment effects in clinical trials. FXS serves as a good model to develop such designs for other developmental disorders, particularly because FXS is a single genetic disorder in which affected individuals all have the same basic cellular defect, a mouse model is available, some information on synaptic function of FMRP in brain is known, and aspects of FXS model more common disorders with likely mechanistic overlap, including ASDs, ADHD, and learning disabilities.
Some lessons that have been learned from early clinical trials completed to date in FXS subjects are as follows. It is very difficult to identify good cognitive outcome measures for drug effect that can be performed by FXS subjects crossing the entire range of function because of problems with basal scores and ceilings (Berry-Kravis et al, 2006, 2009), and more work to validate appropriate measures is needed. Predicting the outcome that will improve most in a complex disorder during initial trials is essentially guesswork, when the treatment targets the underlying disorder and not a specific behavior. Therefore, it is important to run small exploratory trials initially to obtain information from which to choose the primary outcome in subsequent larger trials, and set inclusion criteria to select for subjects with problems in the area targeted by the drug. The factor structure of existing key rating scales such as the ABC (Maltas et al, 2011, personal communication) may not be appropriate for FXS if the measure was developed in a non-FXS cohort. This may prevent detection of improvement in a behavioral area because of dilution of scores with items that are not contributing to that behavior. The placebo effect in FXS trials is large (Berry-Kravis et al, 2011), most likely because many families are aware of current research. Parents who fill out the rating scales meant to detect medication effect are hoping for and expecting improvement from targeted treatments. Thus, findings from short open-label trials may not be reproduced in placebo-controlled trials. A placebo lead-in (period of placebo treatment for all subjects before randomization to active drug or placebo) is a strategy for FXS trials to limit the placebo effect and allow the treatment effect to be more evident. Recruitment is very difficult and prolonged when concomitant medications are not allowed (Erickson et al, 2011a), and thus reasonable concomitant medications that are not predicted to have a major interaction with the agent being studied, will need to be allowed in FXS trials. In fact, this will make a prediction possible about whether the agent being tested can add incrementally to currently existing therapy. Finally, it appears likely that subgroups of patients will respond better to different targeted agents, even in a uniform disorder such as FXS. There may be biomarkers or markers of clinical severity that define a treatment response just in a subset of patients, as in the socially impaired group in the R-baclofen study (Wang et al, 2010b) and the fully methylated group in the AFQ056 study (Jacquemont et al, 2011).
The design and evaluation of outcome measures for trials of targeted treatments in FXS and neurodevelopmental disorders has proven to be a major hurdle. Outcome measures chosen need to test a broad ability range to prevent low or high-functioning individuals from showing ceiling or floor effects, overcome problems with co-operation and variable performance, be shown to be reproducible, and quantify core defects and correlate with quality of life and true functional improvement. Only a subset of outcome measures utilized in recent trials have turned out to fulfill the majority of these criteria (Berry-Kravis et al, 2006, 2008a, 2008b, 2009) suggesting better, FXS-specific measures are needed. Only recently have investigators begun to develop templates to test the feasibility, reproducibility, and validity assessment before using the measure in a trial (Berry-Kravis et al, 2008b; Hessl et al, 2009; Knox and Berry-Kravis, 2009; Farzin et al, 2011). Choice of outcome measures must also balance use of accepted behavioral measures, which are generally caregiver rating scales (such as the ABC), with precedent for use in drug registration/FDA approval versus use of novel measures (Hessl et al, 2009; Knox and Berry-Kravis, 2009; Farzin et al, 2011) that are more quantitative and may objectively measure core phenotypes and electrophysiology (such as eye tracking or PPI). These novel quantitative measures advance treatment science, but have no precedent for registration, do not clearly predict a specific functional outcome, and are often expensive and difficult to run at multiple sites.
FUTURE DIRECTIONS AND CLINICAL IMPLICATIONS
Based on studies in FXS animal models, three major therapeutic strategies in FXS are emerging (summarized in Figure 1): (1) strategies targeting upstream extracellular neurotransmitter receptors, such as metabotropic glutamate, dopamine, and GABA receptors; (2) strategies targeting the intracellular central signal transduction molecules ERK1/2 or PI3K, which integrate and regulate many different signaling pathways, including those stimulating protein synthesis; and (3) strategies targeting multiple further downstream located signaling molecules or proteins translated from FMRP target mRNAs, shown to be dysregulated in FXS (eg, GSK3β, MMP-9, or PAK). Furthermore, particularly promising groups of FMRP-regulated molecules, for example, those involved in AMPA receptor endocytosis (Waung and Huber, 2009), or potassium channels might become candidate therapeutic targets in FXS.
Although some of these strategies are already being tested in clinical trials with patients, future studies in FXS animal models are needed to investigate which (or which combinations) of these therapeutic strategies might be the most beneficial for specific FXS patients, each presenting a slightly different phenotype of the syndrome. Ultimately, the value of these complementary therapeutic strategies will strongly depend on the availability of specific drugs that are safe for use in patients and that restore balance of brain signaling.
Several years ago, the Fragile X Clinical and Research Consortium (FXCRC) was created with the support of CDC funding to help ensure state-of-the-art care delivery to meet the needs of individuals with FXS across the country and facilitate large-scale national research efforts. This organization will also allow FXS Clinics across North America to collaborate in preparation for large multisite clinical trials that will be necessary for FDA approval of targeted treatments and to eventually study combinations of therapies.
It is likely that no one agent will be a successful ‘cure’ for FXS; rather, patients will display optimal responses to different targeted treatments based on individualized complex interactions between genetic variability and neuronal pathway and synaptic function. Thus, it is likely that different patients will function best with certain pathway targets or certain combinations of treatments. There is hope that ongoing development of new targets will gradually build on previous knowledge to result in progressive improvement in treatments to reverse core deficits in all patients with FXS in the future.
Clearly, there is overlap in molecular and synaptic pathways between FXS and autism (Awadalla et al, 2010; Pinto et al, 2010, see Box 1). Thus targeted treatments for FXS will likely also target dysregulated synaptic mechanisms in a subgroup of patients with autism who have defects in the same pathways that are abnormal in FXS. Successful treatments in FXS are expected to be extended to cohorts with ASDs and other cognitive disorders. Progress in development of these targeted treatments for FXS may result, for the first time, in the possibility of medical intervention to reverse CNS defects and resultant clinical manifestations of developmental cognitive disorders and intellectual disability. The exciting progress made thus far and promise for the continued development of therapeutic strategies results from an in-depth analysis of underlying disease mechanisms and availability of animal models to test restoration of brain functions.
References
(The annotated references were selected based on their importance for the development of therapeutic strategies for FXS. They describe: (1) the discovery of FXS phenotypes in animal models that proved to be useful to develop and/or evaluate therapeutic strategies, (2) rescue of FXS-associated phenotypes using either genetic or pharmacologic strategies in animal models, or (3) important clinical trials with FXS patients. The authors wish to note that this is not intended to depreciate the scientific value of the many excellent basic science studies that were not annotated.)
Adusei DC, Pacey LKK, Chen D, Hampson DR (2010). Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology 59: 167–171.
Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci 24: 2648–2655.
Auerbach BD, Bear MF (2010). Loss of the fragile X mental retardation protein decouples metabotropic glutamate receptor dependent priming of long-term potentiation from protein synthesis. J Neurophysiol 104: 1047–1051.
Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR et al (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 87: 316–324.
Banday AA, Fazili FR, Lokhandwala MF (2007). Insulin causes renal dopamine D1 receptor desensitization via GRK2-mediated receptor phosphorylation involving phosphatidylinositol 3-kinase and protein kinase C. Am J Physiol Renal Physiol 293: F877–F884.
Banko JL, Hou L, Poulin F, Sonenberg N, Klann E (2006). Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci 26: 2167–2173.
Bassell GJ, Warren ST (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60: 201–214.
Bear MF, Huber KM, Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci 27: 370–377. Postulation of the ‘mGluR theory of FXS’, which suggests mGlu 1/5 receptors as therapeutic targets for FXS.
Beaulieu J-M, Gainetdinov RR (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63: 182–217.
Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P et al (2009). A novel function for fragile X mental retardation protein in translational activation. PLoS Biol 7: e16.
Béïque J-C, Na Y, Kuhl D, Worley PF, Huganir RL (2011). Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci USA 108: 816–821.
Bernardet M, Crusio WE (2006). Fmr1 KO mice as a possible model of autistic features. ScientificWorldJournal 6: 1164–1176.
Berridge MJ, Downes CP, Hanley MR (1989). Neural and developmental actions of lithium: a unifying hypothesis. Cell 59: 411–419.
Berry-Kravis E (2002). Epilepsy in fragile X syndrome. Dev Med Child Neurol 44: 724–728.
Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW et al (2007). Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord 22: 2018–2030, quiz 2140.
Berry-Kravis E, Grossman AW, Crnic LS, Greenough WT (2002). Understanding fragile X syndrome. Curr Pediatr 12: 316–324.
Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J et al (2009). A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet 46: 266–271. The first treatment of individuals with FXS with an mGlu5 inhibitor: objective improvement in PPI deficits with treatment.
Berry-Kravis E, Knox A, Hervey C (2011). Targeted treatments for fragile X syndrome. J Neurodev Disord, in press (e-pub ahead of print 19 February 2011).
Berry-Kravis E, Krause SE, Block SS, Guter S, Wuu J, Leurgans S et al (2006). Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J Child Adolesc Psychopharmacol 16: 525–540.
Berry-Kravis E, Potanos K (2004). Psychopharmacology in fragile X syndrome—present and future. Ment Retard Dev Disabil Res Rev 10: 42–48.
Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, Bailey DB (2010). Seizures in fragile X syndrome: characteristics and comorbid diagnoses. Am J Intellect Dev Disabil 115: 461–472.
Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N et al (2008a). Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr 29: 293–302.
Berry-Kravis E, Sumis A, Kim OK, Lara R, Wuu J (2008b). Characterization of potential outcome measures for future clinical trials in fragile X syndrome. J Autism Dev Disord 38: 1751–1757.
Bhogal B, Jongens TA (2010). Fragile X syndrome and model organisms: identifying potential routes of therapeutic intervention. Dis Model Mech 3: 693–700.
Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW et al (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 46: 94–102. Rescue of FXS-associated phenotypes in the FXS mouse model with the MMP-9 antagonist minocycline.
Blajecka K, Borgstrom A, Arcaro A (2011). Phosphatidylinositol 3-kinase isoforms as novel drug targets. Curr Drug Targets 12: 1056–1081.
Boccia ML, Roberts JE (2000). Behavior and autonomic nervous system function assessed via heart period measures: the case of hyperarousal in boys with fragile X syndrome. Behav Res Methods Instrum Comput 32: 5–10.
Braden ML (2002). Academic interventions. In: Hagerman RJ, Hagerman PJ (eds). Fragile X Syndrome: Diagnosis, Treatment and Research, 3rd edn. The Johns Hopkins University Press: Baltimore pp 428–464.
Brodkin ES (2008). Social behavior phenotypes in fragile X syndrome, autism, and the Fmr1 knockout mouse: theoretical comment on McNaughton et al. (2008). Behav Neurosci 122: 483–489.
Brosnan J, Healy O (2011). A review of behavioral interventions for the treatment of aggression in individuals with developmental disabilities. Res Dev Disabil 32: 437–446.
Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ et al (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci 13: 819–821.
Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA et al (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107: 477–487.
Bureau I, Shepherd GMG, Svoboda K (2008). Circuit and plasticity defects in the developing somatosensory cortex of Fmr1 knock-out mice. J Neurosci 28: 5178–5188.
Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN et al (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42: 318–321.
Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS et al (2007). Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem 282: 12619–12628.
Centonze D, Rossi S, Napoli I, Mercaldo V, Lacoux C, Ferrari F et al (2007). The brain cytoplasmic RNA BC1 regulates dopamine D2 receptor-mediated transmission in the striatum. J Neurosci 27: 8885–8892.
Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P et al (2008). Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol 4: 256–263. First study showing that GABA can rescue behavioral phenotypes and mushroom body defects in dfmr1 mutant flies.
Chen LY, Rex CS, Babayan AH, Kramar EA, Lynch G, Gall CM et al (2010). Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci 30: 10977–10984.
Choi CH, McBride SM, Schoenfeld BP, Liebelt DA, Ferreiro D, Ferrick NJ et al (2010). Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 11: 347–362.
Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK (2005). Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci 25: 8048–8055.
Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME et al (2010a). Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A 152A: 2512–2520.
Collins SC, Coffee B, Benke PJ, Berry-Kravis E, Gilbert F, Oostra B et al (2010b). Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS One 5: e9476. Two studies identifying novel fmr1 mutations in patients with neurodevelopmental disorders.
Cruz-Martin A, Crespo M, Portera-Cailliau C (2010). Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30: 7793–7803.
Curia G, Papouin T, Séguéla P, Avoli M (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex 19: 1515–1520.
Cusco I, Medrano A, Gener B, Vilardell M, Gallastegui F, Villa O et al (2009). Autism-specific copy number variants further implicate the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder. Hum Mol Genet 18: 1795–1804.
Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR et al (2005). Kissing complex RNAs mediate interaction between the fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev 19: 903–918.
Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107: 489–499.
Davidkova G, Carroll RC (2007). Characterization of the role of microtubule-associated protein 1B in metabotropic glutamate receptor-mediated endocytosis of AMPA receptors in hippocampus. J Neurosci 27: 13273–13278.
Davis E, Saeed S, Antonacci D (2008). Anxiety disorders in persons with developmental disabilities: empirically informed diagnosis and treatment. Psychiatr Q 79: 249–263.
De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B et al (1993). A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet 3: 31–35.
den Broeder MJ, van der Linde H, Brouwer JR, Oostra BA, Willemsen R, Ketting RF (2009). Generation and characterization of Fmr1 knockout Zebrafish. PLoS One 4: e7910.
Deng PY, Xiao Z, Lei S (2010). Distinct modes of modulation of GABAergic transmission by Group I metabotropic glutamate receptors in rat entorhinal cortex. Hippocampus 20: 980–993.
Depositario-Cabacar DFT, Zelleke T-G (2010). Treatment of epilepsy in children with developmental disabilities. Dev Disabil Res Rev 16: 239–247.
De Rubeis S, Bagni C (2010). Fragile X mental retardation protein control of neuronal mRNA metabolism: insights into mRNA stability. Mol Cell Neurosci 43: 43–50.
Desai NS, Casimiro TM, Gruber SM, Vanderklish PW (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol 96: 1734–1745.
De Sarno P, Li X, Jope RS (2002). Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 43: 1158–1164.
de Vries BB, Wiegers AM, Smits AP, Mohkamsing S, Duivenvoorden HJ, Fryns JP et al (1996). Mental status of females with an FMR1 gene full mutation. Am J Hum Genet 58: 1025–1032.
de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL et al (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis 31: 127–132.
D'Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA et al (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res 1121: 238–245.
Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell 14: 926–939.
Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S et al (2007). Correction of fragile X syndrome in mice. Neuron 56: 955–962. Proof of the mGluR theory by genetic rescue of FXS phenotypes in Fmr1 KO mice that were crossed into an mGlu5 heterozygous background.
Eagleson KL, Gravielle MC, Schlueter McFadyen-Ketchum LJ, Russek SJ, Farb DH, Levitt P (2010). Genetic disruption of the autism spectrum disorder risk gene PLAUR induces GABAA receptor subunit changes. Neuroscience 168: 797–810.
Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN et al (2010). Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 65: 373–384.
El Idrissi A, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett 377: 141–146.
Erickson CA, Mullett JE, McDougle CJ (2009). Open-label memantine in fragile X syndrome. J Autism Dev Disord 39: 1629–1635.
Erickson CA, Mullett JE, McDougle CJ (2010). Brief report: acamprosate in fragile X syndrome. J Autism Dev Disord 40: 1412–1416.
Erickson CA, Stigler KA, Wink LK, Mullett JE, Kohn A, Posey DJ et al (2011a). A prospective open-label study of aripiprazole in fragile X syndrome. Psychopharmacology (Berl) 216: 85–90.
Erickson CA, Weng N, Weiler IJ, Greenough WT, Stigler KA, Wink LK et al (2011b). Open-label riluzole in fragile X syndrome. Brain Res 1380: 264–270.
Estes PS, O'Shea M, Clasen S, Zarnescu DC (2008). Fragile X protein controls the efficacy of mRNA transport in Drosophila neurons. Mol Cell Neurosci 39: 170–179.
Ey E, Leblond CS, Bourgeron T (2011). Behavioral profiles of mouse models for autism spectrum disorders. Autism Res 4: 5–16.
Fahling M, Mrowka R, Steege A, Kirschner KM, Benko E, Forstera B et al (2009). Translational regulation of the human achaete-scute homologue-1 by fragile X mental retardation protein. J Biol Chem 284: 4255–4266.
Farzin F, Rivera SM, Hessl D (2009). Brief report: visual processing of faces in individuals with fragile X syndrome: an eye tracking study. J Autism Dev Disord 39: 946–952.
Farzin F, Scaggs F, Hervey C, Berry-Kravis E, Hessl D (2011). Reliability of eye tracking and pupillometry measures in individuals with fragile X syndrome. J Autism Dev Disord, in press (e-pub ahead of print 26 January 2011).
Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST (1997). FMRP associates with polyribosomes as an mRNP and the I304N mutation of severe fragile x syndrome abolishes this association. Mole Cell 1: 109–118.
Gatto CL, Broadie K (2008). Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135: 2637–2648.
Gatto CL, Broadie K (2009). Temporal requirements of the fragile X mental retardation protein in modulating circadian clock circuit synaptic architecture. Front Neural Circuits 5: 12.
Gibson JR, Bartley AF, Hays SA, Huber KM (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol 100: 2615–2626.
Giuffrida R, Musumeci S, D'Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA et al (2005). A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J Neurosci 25: 8908–8916.
Gladding CM, Fitzjohn SM, Molnar E (2009). Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol Rev 61: 395–412.
Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K et al (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci 30: 10624–10638. Evidence that a PI3K antagonist can rescue FXS phenotypes in Fmr1 KO mice.
Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 31: 5693–5698.
Grossman AW, Aldridge GM, Lee KJ, Zeman MK, Jun CS, Azam HS et al (2010). Developmental characteristics of dendritic spines in the dentate gyrus of Fmr1 knockout mice. Brain Res 1355: 221–227.
Hagerman RJ (2002). Physical and behavioral phenotype. In: Hagerman RJ, Hagerman PJ (eds). Fragile X Syndrome: Diagnosis, Treatment and Research, 3rd edn. The Johns Hopkins University Press: Baltimore.
Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A et al (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123: 378–390.
Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A (2010). Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65: 385–398.
Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY et al (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA 104: 11489–11494. Description of a genetic rescue in Fmr1 KO mice by overexpression of dominant-negative PAK, which initiated efforts to develop PAK-specific antagonists for treatment of individuals with FXS.
Hessl D, Berry-Kravis E, Cordeiro L, Yuhas J, Ornitz EM, Campbell A et al (2009). Prepulse inhibition in fragile X syndrome: feasibility, reliability, and implications for treatment. Am J Med Genet B Neuropsychiatr Genet 150B: 545–553.
Hessl D, Tassone F, Cordeiro L, Koldewyn K, McCormick C, Green C et al (2008). Brief report: aggression and stereotypic behavior in males with fragile X syndrome--moderating secondary genes in a “single gene” disorder. J Autism Dev Disord 38: 184–189.
Heulens I, D'Hulst C, Braat S, Rooms L, Kooy RF (2010). Involvement and therapeutic potential of the GABAergic system in the fragile X syndrome. ScientificWorldJournal 10: 2198–2206.
Hirono M, Yoshioka T, Konishi S (2001). GABA(B) receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat Neurosci 4: 1207–1216.
Hoeft F, Hernandez A, Parthasarathy S, Watson CL, Hall SS, Reiss AL (2007). Fronto-striatal dysfunction and potential compensatory mechanisms in male adolescents with fragile X syndrome. Hum Brain Mapp 28: 543–554.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51: 441–454.
Huber KM (2006). The fragile X-cerebellum connection. Trends Neurosci 29: 183–185.
Huber KM, Gallagher SM, Warren ST, Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile x mental retardation. Proc Natl Acad Sci USA 99: 7746–7750. Seminal study that showed exaggerated mGlu1/5-dependent LTD in Fmr1 KO hippocampus and motivated the mGluR theory of FXS.
Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ et al (2002). Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet 111: 140–146. Description of abnormal dendritic spine morphology in the FXS mouse model due to deletion of FMRP.
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP et al (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98: 161–167.
Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ et al (2011). Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med 3: 64ra61. First placebo-controlled clinical trial of an mGlu5 blocker that showed improvement in a group with full methylation of FMR1.
Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA et al (2004). Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci 7: 113–117.
Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M (2009). Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci 29: 8688–8697.
Kelleher III RJ, Bear MF (2008). The autistic neuron: troubled translation? Cell 135: 401–406.
Kim SH, Markham JA, Weiler IJ, Greenough WT (2008). Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci USA 105: 4429–4434.
Knox A, Berry-Kravis E (2009). Feasibility and reproducibility of the Tests of Attentional Performance for Children (KiTAP) in Fragile X Syndrome (FXS). Ann Neurol 66: S108.
Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R et al (2005). Deletion of FMR1 in purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in fragile X syndrome. Neuron 47: 339–352.
Koishi S, Yamamoto K, Matsumoto H, Koishi S, Enseki Y, Oya A et al (2006). Serotonin transporter gene promoter polymorphism and autism: a family-based genetic association study in Japanese population. Brain Dev 28: 257–260.
Kolaj M, Renaud LP (2010). Metabotropic glutamate receptors in median preoptic neurons modulate neuronal excitability and glutamatergic and GABAergic inputs from the subfornical organ. J Neurophysiol 103: 1104–1113.
Kolasiewicz W, Kuter K, Wardas J, Ossowska K (2009). Role of the metabotropic glutamate receptor subtype 1 in the harmaline-induced tremor in rats. J Neural Transm 116: 1059–1063.
Kronk R, Bishop EE, Raspa M, Bickel JO, Mandel DA, Bailey Jr DB (2010). Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep 33: 679–687.
Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF (2011). Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA 108: 2587–2592. Suggests that defects in prefrontal synaptic plasticity in FXS mice might be beneficial to evaluate novel therapeutic strategies for FXS.
Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y et al (1997). Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem 272: 24252–24256.
Larson J, Jessen RE, Kim D, Fine AK, du Hoffmann J (2005). Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci 25: 9460–9469.
Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G et al (2007). Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci 27: 10685–10694.
Lebel M, Patenaude C, Allyson J, Massicotte G, Cyr M (2009). Dopamine D1 receptor activation induces tau phosphorylation via cdk5 and GSK3 signaling pathways. Neuropharmacology 57: 392–402.
Levenga J, de Vrij FMS, Oostra BA, Willemsen R (2010). Potential therapeutic interventions for fragile X syndrome. Trends Mol Med 16: 516–527.
Levitt P, Campbell DB (2009). The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest 119: 747–754.
Li C, Bassell GJ, Sasaki Y (2009). Fragile X mental retardation protein is involved in protein synthesis-dependent collapse of growth cones induced by semaphorin-3A. Front Neural Circuits 3: 11.
Li J, Pelletier MR, Perez Velazquez JL, Carlen PL (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci 19: 138–151.
Lim L, Manser E, Leung T, Hall C (1996). Regulation of phosphorylation pathways by p21 GTPases. Eur J Biochem 242: 171–185.
Liu Z-H, Chuang D-M, Smith CB (2011). Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 14: 618–630.
Lu R, Wang H, Liang Z, Ku L, O'Donnell WT, Li W et al (2004). The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci USA 101: 15201–15206.
Maddalena A, Richards CS, McGinniss MJ, Brothman A, Desnick RJ, Grier RE et al (2001). Technical standards and guidelines for fragile X: the first of a series of disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med 3: 200–205.
Maino DM, Wesson M, Schlange D, Cibis G, Maino JH (1991). Optometric findings in the fragile X syndrome. Optom Vis Sci 68: 634–640.
McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D et al (2005). Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45: 753–764. Proof of the mGluR theory in a Drosophila model for FXS shows that mGlu antagonists and lithium can rescue FXS-associated phenotypes in dfmr1 mutant Drosophila.
McKinney BC, Grossman AW, Elisseou NM, Greenough WT (2005). Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am J Med Genet B Neuropsychiatr Genet 136B: 98–102.
Mercaldo V, Descalzi G, Zhuo M (2009). Fragile X mental retardation protein in learning-related synaptic plasticity. Mol Cells 28: 501–507.
Meredith RM, Holmgren CD, Weidum M, Burnashev N, Mansvelder HD (2007). Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54: 627–638.
Miao Y, Zhang Y, Wan H, Chen L, Wang F (2010). GABA-receptor agonist, propofol inhibits invasion of colon carcinoma cells. Biomed Pharmacother 64: 583–588.
Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK et al (1999). Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet 83: 268–279.
Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS et al (2009). Elevated glycogen synthase kinase-3 activity in fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56: 463–472. Rescue study in the FXS mouse model with lithium, which motivated clinical trials in FXS with the GSK3 β inhibitor lithium.
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One 5: e9706.
Minichiello L (2009). TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10: 850–860.
Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L et al (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37: 417–431.
Morales J, Hiesinger PR, Schroeder AJ, Kume K, Verstreken P, Jackson FR et al (2002). Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34: 961–972.
Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci 27: 5338–5348.
Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O et al (2011). Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation and mGluR signaling. Mol Cell 42: 673–688.
Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M et al (2000). Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41: 19–23. First report describing the audiogenic seizure phenotype in FXS mice.
Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA et al (1999). Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40: 1092–1099.
Nagai T, Takuma K, Kamei H, Ito Y, Nakamichi N, Ibi D et al (2007). Dopamine D1 receptors regulate protein synthesis-dependent long-term recognition memory via extracellular signal-regulated kinase 1/2 in the prefrontal cortex. Learn Mem 14: 117–125.
Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci USA 104: 15537–15542. First description of a role of FMRP for GluA internalization, and rescue of this phenotype by MPEP.
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S et al (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134: 1042–1054.
Nimchinsky EA, Oberlander AM, Svoboda K (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci 21: 5139–5146.
Nosyreva ED, Huber KM (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95: 3291–3295. Important follow-up study to the first report on dysregulated LTD in FXS (Huber et al., 2002), showing that LTD in Fmr1 KO mice is protein synthesis independent.
Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci 30: 9929–9938. Shows rescue of impaired neuronal function in the amygdala of FXS mice with a GABA agonist.
Osterweil EK, Krueger DD, Reinhold K, Bear MF (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 30: 15616–15627. In vivo rescue of seizures in Fmr1 KO mice with an ERK1/2 inhibitor.
Pacey LK, Doss L, Cifelli C, der Kooy DV, Heximer SP, Hampson DR (2011). Genetic deletion of regulator of G-protein signaling 4 (RGS4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol Cell Neurosci 46: 563–572. Evidence from mouse studies that RGS4 might be a promising therapeutic target in FXS.
Pan F, Aldridge GM, Greenough WT, Gan WB (2010). Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA 107: 17768–17773.
Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94: 185–192.
Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell I et al (2010). Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol 10: 91.
Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL et al (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59: 70–83.
Peebles CL, Yoo J, Thwin MT, Palop JJ, Noebels JL, Finkbeiner S (2010). Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci USA 107: 18173–18178.
Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14: 285–293.
Pfeiffer BE, Huber KM (2007). Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J Neurosci 27: 3120–3130.
Pfeiffer BE, Huber KM (2009). The state of synapses in fragile X syndrome. Neuroscientist 15: 549–567.
Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN et al (2010). Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66: 191–197.
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT et al (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66: 817–822.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R et al (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466: 368–372.
Portera-Cailliau C (2011). Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity? Neuroscientist, in press (e-pub ahead of print 6 May 2011).
Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F (2007). Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci 27: 13958–13967.
Qin M, Kang J, Burlin TV, Jiang C, Smith CB (2005). Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the Fmr1 null mouse. J Neurosci 25: 5087–5095.
Qiu L-F, Lu T-J, Hu X-L, Yi Y-H, Liao W-P, Xiong Z-Q (2009). Limbic epileptogenesis in a mouse model of fragile X syndrome. Cereb Cortex 19: 1504–1514.
Raspa M, Bailey DB, Bishop E, Holiday D, Olmsted M (2010). Obesity, food selectivity, and physical activity in individuals with fragile X syndrome. Am J Intellect Dev Disabil 115: 482–495.
Redfern RE, Daou M-C, Li L, Munson M, Gericke A, Ross AH (2010). A mutant form of PTEN linked to autism. Protein Sci 19: 1948–1956.
Reiss AL, Hall SS (2007). Fragile X syndrome: assessment and treatment implications. Child Adolesc Psychiatr Clin N Am 16: 663–675.
Ronesi JA, Huber KM (2008). Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci 28: 543–547.
Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA et al (2003). PI3 kinase enhancer-homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci 6: 1153–1161.
Santos AR, Comprido D, Duarte CB (2010). Regulation of local translation at the synapse by BDNF. Prog Neurobiol 92: 505–516.
Schicknick H, Schott BH, Budinger E, Smalla K-H, Riedel A, Seidenbecher CI et al (2008). Dopaminergic modulation of auditory cortex-dependent memory consolidation through mTOR. Cereb Cortex 18: 2646–2658.
Schultz-Pedersen S, Hasle H, Olsen JH, Friedrich U (2001). Evidence of decreased risk of cancer in individuals with fragile X. Am J Med Genet 103: 226–230.
Schutt J, Falley K, Richter D, Kreienkamp HJ, Kindler S (2009). Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem 284: 25479–25487.
Scotto-Lomassese S, Nissant A, Mota T, Neant-Fery M, Oostra BA, Greer CA et al (2011). Fragile X mental retardation protein regulates new neuron differentiation in the adult olfactory bulb. J Neurosci 31: 2205–2215.
Shang Y, Wang H, Mercaldo V, Li X, Chen T, Zhuo M (2009). Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J Neurochem 111: 635–646.
Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E et al (2010). Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 30: 694–702. Report showing dysregulated mTOR signaling downstream of mGlu 1/5 in the absence of FMRP, suggesting mTOR as a potential therapeutic target in FXS.
Sheikh AM, Malik M, Wen G, Chauhan A, Chauhan V, Gong C-X et al (2010). BDNF-Akt-Bcl2 antiapoptotic signaling pathway is compromised in the brain of autistic subjects. J Neurosci Res 88: 2641–2647.
Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D et al (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52: 475–484.
Sherman SL (2000). Premature ovarian failure in the fragile X syndrome. Am J Med Genet 97: 189–194.
Silverman JL, Yang M, Lord C, Crawley JN (2010). Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11: 490–502.
Soden ME, Chen L (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci 30: 16910–16921.
Spencer CM, Alekseyenko O, Hamilton SM, Thomas AM, Serysheva E, Yuva-Paylor LA et al (2011). Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res 4: 40–56.
Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R (2005). Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav 4: 420–430.
Steckler T, Oliveira AF, Van Dyck C, Van Craenendonck H, Mateus AM, Langlois X et al (2005). Metabotropic glutamate receptor 1 blockade impairs acquisition and retention in a spatial Water maze task. Behav Brain Res 164: 52–60.
Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK (2010). Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci 30: 10263–10271.
Sullivan K, Hooper S, Hatton D (2007). Behavioural equivalents of anxiety in children with fragile X syndrome: parent and teacher report. J Intellect Disabil Res 51: 54–65.
Sund R, Pukkala E, Patja K (2009). Cancer incidence among persons with fragile X syndrome in Finland: a population-based study. J Intellect Disabil Res 53: 85–90.
Suvrathan A, Chattarji S (2011). Fragile X syndrome and the amygdala. Curr Opin Neurobiol, in press (e-pub ahead of print 7 May 2011).
Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S (2010). Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci 107: 11591–11596.
Tessier CR, Broadie K (2008). Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135: 1547–1557.
The Dutch-Belgian Fragile XC, Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F et al (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78: 23–33. Generation and description of the Fmr1 knockout mouse, which became an indispensable tool for development and test of therapeutic strategies for FXS.
Todd PK, Malter JS, Mack KJ (2003). Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res 110: 267–278.
Tucker B, Richards RI, Lardelli M (2006). Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum Mol Genet 15: 3446–3458.
Tyagi N, Gillespie W, Vacek JC, Sen U, Tyagi SC, Lominadze D (2009). Activation of GABA-A receptor ameliorates homocysteine-induced MMP-9 activation by ERK pathway. J Cell Physiol 220: 257–266.
Utari A, Chonchaiya W, Rivera SM, Schneider A, Hagerman RJ, Faradz SM et al (2010). Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am J Intellect Dev Disabil 115: 433–443.
Van Dam D, D'Hooge R, Hauben E, Reyniers E, Gantois I, Bakker CE et al (2000). Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav Brain Res 117: 127–136.
van de Lagemaat LN, Grant SG (2010). Genome variation and complexity in the autism spectrum. Neuron 67: 8–10.
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A et al (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914.
Volk LJ, Pfeiffer BE, Gibson JR, Huber KM (2007). Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci 27: 11624–11634. Expansion of the mGluR theory showing that signaling downstream of other Gq-coupled receptors is also dysregulated in Fmr1 KO.
Wang H, Fukushima H, Kida S, Zhuo M (2009). Ca2+/calmodulin-dependent protein kinase IV links group I metabotropic glutamate receptors to fragile X mental retardation protein in cingulate cortex. J Biol Chem 284: 18953–18962.
Wang H, Kim SS, Zhuo M (2010a). Roles of fragile X mental retardation protein in dopaminergic stimulation-induced synapse-associated protein synthesis and subsequent alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-4-propionate (AMPA) receptor internalization. J Biol Chem 285: 21888–21901.
Wang H, Wu LJ, Kim SS, Lee FJ, Gong B, Toyoda H et al (2008b). FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 59: 634–647. First report showing dysregulated dopamine receptor signaling in FXS mice.
Wang H, Wu LJ, Zhang F, Zhuo M (2008a). Roles of calcium-stimulated adenylyl cyclase and calmodulin-dependent protein kinase IV in the regulation of FMRP by group I metabotropic glutamate receptors. J Neurosci 28: 4385–4397.
Wang LW, Berry-Kravis E, Hagerman RJ (2010b). Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics 7: 264–274.
Waung MW, Huber KM (2009). Protein translation in synaptic plasticity: mGluR-LTD, fragile X. Curr Opin Neurobiol 19: 319–326.
Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM (2008). Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59: 84–97.
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Eberwine J, Greenough WT (1997). Fragile X mental retardation protein is translated near synapses. Proc Natl Acad Sci USA 94: 5395–5400.
Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V et al (2004). Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA 101: 17504–17509.
Welch HC, Coadwell WJ, Stephens LR, Hawkins PT (2003). Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett 546: 93–97.
Weng N, Weiler IJ, Sumis A, Berry-Kravis E, Greenough WT (2008). Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am J Med Genet B Neuropsychiatr Genet 147B: 1253–1257.
Wiznitzer M (2004). Autism and tuberous sclerosis. J Child Neurol 19: 675–679.
Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP (2004). A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav 3: 337–359.
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP (2005). Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49: 1053–1066. First pharmacologic proof of the mGluR theory showing that MPEP rescues FXS-related phenotypes in Fmr1 KO mice.
Yoshii A, Constantine-Paton M (2010). Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70: 304–322.
Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS (2010). Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol 79: 632–646.
Zalfa F, Achsel T, Bagni C (2006). mRNPs, polysomes or granules: FMRP in neuronal protein synthesis. Curr Opin Neurobiol 16: 265–269.
Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A et al (2007). A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci 10: 578–587.
Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S et al (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112: 317–327.
Zhang L, Alger BE (2010). Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome. J Neurosci 30: 5724–5729.
Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD et al (2001). Drosophila Fragile X-related gene regulates the MAP1b homolog Futsch to control synaptic structure and function. Cell 107: 591–603.
Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M (2005). Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci 25: 7385–7392.
Zhou X, Lin DS, Zheng F, Sutton MA, Wang H (2010). Intracellular calcium and calmodulin link brain-derived neurotrophic factor to p70S6 kinase phosphorylation and dendritic protein synthesis. J Neurosci Res 88: 1420–1432.
Acknowledgements
We thank Sharon Swanger for helpful discussions and critically reading the manuscript. This work was supported by a postdoctoral fellowship and Conquer Fragile X research grant from the National Fragile X Foundation (to CG), the NIH Grant MH085617 (to GJB), and the Fragile X Center Grant 3P30HD024064 (to GJB).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
EMB-K has served as a consultant and has received funding for clinical trials from Neuropharm LTD, Seaside Therapeutics, Novartis Pharmaceuticals, and Roche Pharmaceuticals. GJB and CG declare no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Gross, C., Berry-Kravis, E. & Bassell, G. Therapeutic Strategies in Fragile X Syndrome: Dysregulated mGluR Signaling and Beyond. Neuropsychopharmacol 37, 178–195 (2012). https://doi.org/10.1038/npp.2011.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.137
Keywords
This article is cited by
-
Deletion of Fmr1 in parvalbumin-expressing neurons results in dysregulated translation and selective behavioral deficits associated with fragile X syndrome
Molecular Autism (2022)
-
Fragile X mental retardation protein in intrahepatic cholangiocarcinoma: regulating the cancer cell behavior plasticity at the leading edge
Oncogene (2021)
-
Pharmacological Rescue of Hippocampal Fear Learning Deficits in Fragile X Syndrome
Molecular Neurobiology (2018)
-
The fragile X mental retardation protein regulates tumor invasiveness-related pathways in melanoma cells
Cell Death & Disease (2017)
-
Challenges in Conducting Clinical Trials for Pharmacotherapies in Fragile X Syndrome: Lessons Learned
Pharmaceutical Medicine (2017)
