
Abstract
Brown adipose tissue (BAT) is a specialized thermoregulatory organ that has a critical role in the regulation of energy metabolism. Specifically, energy expenditure can be enhanced by the activation of BAT function and the induction of a BAT-like catabolic phenotype in white adipose tissue (WAT). Since the recent recognition of metabolically active BAT in adult humans, BAT has been extensively studied as one of the most promising targets identified for treating obesity and its related disorders. In this review, we summarize information on the developmental origin of BAT and the progenitors of brown adipocytes in WAT. We explore the transcriptional control of brown adipocyte differentiation during classical BAT development and in WAT browning. We also discuss the neuronal control of BAT activity and summarize the recently identified non-canonical stimulators of BAT that can act independently of β-adrenergic stimulation. Finally, we review new findings on the beneficial effects of BAT activation and development with respect to improving metabolic profiles. We highlight the therapeutic potential of BAT and its future prospects, including pharmacological intervention and cell-based therapies designed to enhance BAT activity and development.
Similar content being viewed by others


Introduction
Obesity is a multidimensional metabolic disorder associated with an increased risk of several common and severe diseases, including insulin resistance, type 2 diabetes, cardiovascular diseases and certain types of cancer.1 Recent epidemiological studies have estimated that obesity is a leading contributor to overall disease burden2, 3 and have emphasized obesity’s life-threatening complications. Obesity results from an imbalance that develops between energy intake and expenditure4 when adipose tissue undergoes hypertrophic/hyperplastic expansion during energy oversupply. In many cases, hypertrophied adipose tissue cannot fulfill its storage function, and this exposes excess free fatty acids (FFAs) to lipid-intolerant organs. Consequently, lipotoxicity can induce an insulin-resistant state and low-grade inflammation in the adipose tissue, which can lead to the pathogenesis of metabolic disease.5
Adipose tissue has been traditionally subclassified into white adipose tissue (WAT) and brown adipose tissue (BAT).6 WAT has evolved to store excess energy and to mobilize FFAs when energy is needed. By contrast, BAT is a specialized thermoregulatory organ7 that can produce heat during non-shivering thermogenesis in response to cold exposure. Importantly, energy expenditure can be enhanced by activating the catabolic phenotype of BAT. The recent identification of BAT in adult humans has renewed the interest in the therapeutic use of BAT to treat metabolic diseases.8, 9 One therapeutic strategy involves augmenting the oxidative metabolism of BAT, which has been shown to regulate body weight and temperature.10, 11, 12 In this regard, β3-adrenergic receptor (ADRB3) agonists have been reported to stimulate thermogenesis, increase metabolic rate, and exert anti-obesity and anti-diabetic effects in rodent models.13, 14, 15, 16, 17, 18 Another attractive approach may be to potentially improve the metabolic characteristics of WAT by recruiting brown-like adipocytes, a phenomenon referred to as the browning of WAT.19, 20
In this review, we summarize information published on the origin of brown adipocytes during the development of classical BAT and the browning of WAT, focusing on new advances in our understanding of the identity of brown adipocyte progenitors. We discuss the neuronal and hormonal control of BAT activity and summarize the non-canonical contributors to BAT activity, which increase the thermoregulatory energy expenditure of BAT independently of β-adrenergic stimulation. We also touch upon the transcriptional control of brown adipocyte differentiation that occurs in classical BAT development and WAT browning and review recent findings regarding the beneficial effects of BAT activation on improving metabolic profiles. Finally, we discuss the potential of using pharmacological intervention and cell-based therapies to enhance BAT activity and development, which may have clinical implications for metabolic disease.
Brown adipocyte progenitors
BAT develops prenatally in most mammals and can be easily identified at birth.7 The thermoregulatory function of BAT protects organisms from a cold environment and helps maintain homeostatic body temperature. BAT exists in defined anatomical regions, with the main BAT depots being interscapular, axillary, perirenal and periaortic depots in rodents.7 Human BAT is also currently accepted to be detected in specific anatomical locations such as cervical, supraclavicular and paraspinal regions, which is similar to the BAT distribution in rodents.21 In addition to its histology-based identification, metabolically active BAT in humans can be detected using an imaging technology such as fluoro-deoxyglucose positron emission tomography scanning. Whereas constitutive BAT is found in specific anatomical locations, brown adipocytes can be recruited as clusters that form within WAT depots. These inducible brown adipocytes have a cellular origin that is distinct from that of constitutive brown adipocytes found in classical BAT; however, the identity of brown adipocyte progenitors in various anatomical locations and under physiological conditions remains to be established.
Progenitors of constitutive brown adipocytes
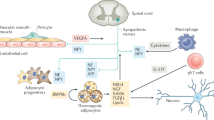
In vivo lineage-tracing studies have demonstrated that cells expressing myogenic factor 5 (Myf5) give rise to skeletal myocytes and brown adipocytes in interscapular and perirenal depots (Figure 1).22 Moreover, lineage tracing performed using an inducible paired box 7 (Pax7) reporter demonstrated that the developmental divergence between BAT and muscles occurs around embryonic day 10.5 (E10.5)–E12.5, supporting the myogenic origin of BAT. Interestingly, inducible brown adipocytes that appear in WAT upon ADRB3 stimulation were not tagged by the Myf5 reporter, indicating that the origins of inducible brown adipocytes are distinct in WAT. However, the concept that brown adipocytes originate in the muscle might be an oversimplification. The Myf5 promoter has been reported to be transiently activated in the undifferentiated paraxial mesoderm, as demonstrated by the expression of the Myf5 reporter in multiple cell types, cartilage and bone.23 Furthermore, recent findings demonstrated that Myf5-expressing progenitors can give rise to white adipocytes in WAT depots.24 Conditional genetic knockout of phosphatase and tensin homolog (PTEN) in Myf5-expressing cells caused a partial lipodystrophic phenotype, suggesting that Myf5+ progenitors contribute in a region-specific manner to WAT depots, which may explain the heterogeneity in the metabolic phenotypes of distinct adipose tissue depots. Although Myf5 expression could be transcriptionally critical in brown adipogenesis, Myf5 expression is highly likely to only indicate the developmental timing of adipose tissue specification, because other mesenchymal cell types also appear to originate from Myf5+ cells. Thus, Myf5 may be considered as an early embryological marker of the paraxial mesoderm. Similarly, lineage tracing performed using Engrailed-1, a central-dermomyotome marker, also demonstrated that interscapular BAT, epaxial muscle and dermis all arise from this embryonic region.25

Cellular origins of brown adipocytes. At least three cellular origins of brown adipocytes exist, all featuring distinct developmental stages and anatomical locations. (1) During developmental growth, constitutive brown adipocytes are generated from Myf5+ cells, the common progenitors of brown adipocytes and myocytes in skeletal muscle. (2) Beige adipocytes found in gonadal WAT originate from PDGFRα+ progenitors, which have been identified as the common progenitors of white and brown adipocytes. (3) Mature white adipocytes can be converted into beige adipocytes in subcutaneous WAT during cold stress.
Brown adipocytes also exist in perivascular fat. Thoracic perivascular depots resemble classical BAT, whereas abdominal perivascular adipose tissue resembles WAT, indicating the heterogeneous metabolic characteristics of perivascular fat.26 Although most perivascular depots remain to be examined, lineage tracing showed that Cre-linked transgelin (smooth muscle protein 22-alpha), which is active in vascular smooth muscle cells and the surrounding mesenchyme, marks perivascular BAT, indicating that the lineages of perivascular BAT and vascular smooth muscle cells are closely related.27, 28
Progenitors of inducible brown adipocytes (brite/beige cells)
Brown adipocytes can be recruited in classical WAT during cold exposure and through adrenergic stimulation.11, 29, 30 Studies on their molecular and metabolic characteristics identified these inducible brown adipocytes as a distinct type of adipocytes that is neither brown nor white.31 Currently, these inducible brown adipocytes are referred to as brown-in-white (‘brite’) or beige adipocytes.32, 33, 34 Although the identity and the origin of inducible brown adipocytes have been defined under distinct circumstances by multiple groups, at least two mechanisms are known to be involved in recruitment of brown adipocytes in WAT: (1) the conversion of mature white adipocytes into brown adipocytes and (2) the de novo generation of brown adipocytes from progenitor cells (Figure 1). The conversion from white adipocytes into brown adipocytes appears to occur predominantly in subcutaneous depots, a view supported by the morphological analyses of brite/beige cells at the ultrastructural levels.35, 36, 37 The browning of white adipocytes has been reported to be a bidirectional conversion, which was demonstrated by the lineage tracing of beige cells in an inducible brown-adipose uncoupling protein 1 (UCP1) reporter model.34 The observation that most UCP1+ cells did not originate from proliferating cells in inguinal adipose tissue also indirectly supports the theory of transdifferentiation.38
The prevalence of inducible brown adipocyte recruitment from proliferating progenitors appears to be depot specific and stimulus dependent. The Granneman group identified a cell type that can become brown or white adipocytes depending on the nature of the stimulus.38 These progenitor cells reside in adipose tissues, express mesenchymal stem cell markers (CD34, Sca1 and PDGFRα),39, 40 and exhibit a unique dendritic morphology. PDGFRα+ cells in gonadal WAT are more prone to be activated by β3-adrenergic stimulation and nutritional challenges. These observations suggest that local tissue-derived signals may control progenitor behavior and lead to depot-specific progenitor recruitment in vivo. Subsequently, follow-up studies performed by Granneman and colleagues demonstrated that M2 macrophages are involved in activating these progenitor cells.41 The recruitment of M2 macrophages was specific to gonadal WAT during β-adrenergic stimulation, and the CD44+ subfraction of PDGFRα+ progenitors differentiated at the site of WAT death. Several adipogenic stimuli, including the feeding of a high fat diet and local adipose-tissue injury, triggered the recruitment of macrophages exhibiting distinct molecular phenotypes and induced the differentiation of PDGFRα+ cells.
Spiegelman et al. characterized the molecular signature of cloned beige-cell progenitor lines generated from the stromovascular faction of subcutaneous adipose tissue.32 In this study, novel markers specific to beige cells (CD137, TEME26 and TBX1) were identified.32 The genetic profiling of human BAT in this report demonstrated that human BAT expresses beige cell markers and not classical BAT markers; however, more detailed expression profiling studies have further dissected the heterogeneous molecular characteristics of human-neck BAT.42, 43 These studies have demonstrated that BAT displays molecular stratification, with deep BAT depots resembling classical BAT and superficial ones expressing beige markers.43
Transcriptional control of brown adipocyte development
Multiple transcriptional regulatory cascades have been reported to control BAT development and thermogenic function (Figure 2).20, 44, 45, 46 Peroxisome proliferator activated receptor gamma (PPARγ) is a master transcription factor that is unconditionally required for white and brown adipogenesis; however, the transcriptional activity of PPARγ can be regulated at multiple levels, enabling brown adipocyte-specific target gene expression.47 Post-transcriptional modifications such as phosphorylation, acetylation, and sumoylation are involved in the control of PPARγ activity. For example, sirtuin1 (Sirt1) deacetylates PPARγ and thereby recruits PRDM16 (PRD1-F1-RIZ1 homologous domain containing 16) to the promoter regions of brown adipocyte-specific target genes, which subsequently stimulates brown adipocytes development.48 Conversely, indirect regulation of PPARγ transcriptional activity includes the differential expression of co-regulators (that is, co-activators and co-repressors).49 Recently, early B cell factor-2 was reported to be more abundantly expressed in BAT than in WAT. EBF2 interacts with PPARγ to specifically activate the BAT developmental program.50 Furthermore, dynamic histone and DNA modifications at the cis-regulatory level can explain depot-specific expression patterns of PPARγ target genes.51 For example, the distinct status of chromatin in BAT and in subcutaneous and gonadal WAT contributes to the depot-selective interaction of PPARγ with PPAR-response elements in different adipocyte lineages.52 Together, these findings illustrate how coordinated regulation at multiple layers enables the fine-tuning of PPARγ transcriptional activity in BAT versus WAT.

Molecular players in brown adipocyte development and function. Brown adipogenic differentiation involves multiple transcriptional regulators, with PRDM16, PPRAγ and PGC1α acting as main molecular players. PPARγ is the master transcription factor in adipogenic differentiation, whose association with PRDM16 and PGC1α initiates selective induction of brown adipogenic and thermogenic gene expression. Acetylation of PPARγ and phosphorylation of PGC1α increase the activity of these transcriptional complexes. Several binding partners of PRDM16 have been identified (CCAAT/enhancer-binding protein beta, PPARγ, early B cell factor-2, euchromatic histone-lysine N-methyltransferase 1 and C-terminal-binding protein) that are involved in mediating the expression of brown adipogenic genes and suppressing the expression of myogenic and white adipogenic genes. Upon sympathetic stimulation, catecholamines (for example, norepinephrine) induce thermogenic gene expression and lipolysis mainly through ADRB-cAMP-PKA signaling. Non-canonical stimulators (for example, natriuretic peptide, BMPs, fibroblast growth factor 21) have also been identified that have critical roles in BAT development and in the browning of WAT (see text for details; main references include).46, 70, 95
PPARγ co-activator 1 alpha (PGC1α) has been identified as a cold-inducible transcriptional cofactor that is involved in thermogenic gene expression leading to, for example, mitochondrial biogenesis, beta-oxidation and lipid metabolism.53 Although PGC1α functions as a co-activator of PPARγ, it is a downstream target gene of the protein kinase A (PKA)-cAMP (cyclic AMP)-responsive element-binding protein 1 signaling pathways, and the transcriptional activity of PGC1α can be post-transcriptionally controlled. Specifically, β-adrenergic stimulation activates p38 mitogen-activated protein kinase, which phosphorylates and activates PGC1α.54
PRDM16 has been identified as a molecular switch between myocytes and brown adipocytes,22 and it is considered as a classical brown adipocyte lineage marker. Whereas the overexpression of PRDM16 converted myocytes into brown adipocytes, PRDM16 expression in adipose tissue specifically induced thermogenic gene expression and browning of subcutaneous WAT, increased energy expenditure and antagonized weight gain during high fat diet feeding.55, 56 Mechanistic studies conducted by multiple groups have demonstrated that PRDM16 forms a transcriptional complex by binding to other transcriptional regulators (Figure 2). For example, CCAAT/enhancer-binding protein beta has been identified as an early binding partner that PRDM16 interacts with in preadipocytes and myogenic precursors to induce PPARγ and PGC1α expression.57 Subsequently, PRDM16 binds to PPARγ and PGC1α to selectively recruit the PPARγ transcription complex to the promoters of thermogenic and brown adipogenic genes.57 Conversely, the suppression of white adipogenic gene expression is mediated by the association of PRDM16 with C-terminal-binding protein-1 and -2, which are well-known transcriptional co-repressors.58 Recent work by Kajimura and colleagues indicated that myogenic gene expression is repressed by the association of PRDM16 with euchromatic histone-lysine N-methyltransferase 1.59 Interestingly, euchromatic histone-lysine N-methyltransferase 1 could stabilize the PRDM16 protein and thereby upregulate brown adipogenic gene expression. Transducin-like enhancer of split 3, a white adipocyte-specific transcription cofactor, competes with PRDM16 and activates anabolic gene expression.60 By contrast, miRNA-133 directly targets PRDM16 expression: downregulation of miRNA-133 upon cold exposure causes augmented expression of PRDM16, resulting in brown adipocyte differentiation into BAT and subcutaneous adipose tissue depots.61 Similarly, reduction of miRNA-133 expression in satellite cells in muscle has been shown to direct brown adipocyte differentiation,62 increase WAT browning and improve metabolic profiles, thus supporting a potential therapeutic use of miRNA-133 in metabolic diseases.63 Although a critical role of PRDM16 in brown adipocyte development and WAT browning is well supported, it would be interesting to test whether cell type-specific manipulation of PRDM16, such as genetic deletion in Myf5+ cells and/or in PDGFRα+ cells, affects the development and metabolic characteristics of BAT and WAT.
Activators of brown adipocyte development and function
Under physiological conditions, cold exposure increases the thermogenic activity of BAT, which is largely controlled by the sympathetic nervous system. Increased sympathetic nerve activity can be detected following cold exposure, which is demonstrated by elevated levels of tyrosine hydroxylase expression, increased nerve fiber branching and norepinephrine turnover.64, 65, 66
Norepinephrine, which is released from sympathetic nerve endings, acts on ADRBs in BAT and activates downstream cAMP-PKA signaling to increase thermoregulatory gene expression and lipolysis through the phosphorylation of hormone-sensitive lipase.
Thermogenesis is critically affected by UCP1, a brown adipocyte-specific molecular marker. UCP1 can be found in the mitochondrial inner membrane, where it uncouples oxidative respiration from ATP synthesis to generate heat. As mentioned in the preceding section, another critical factor upregulated by PKA is PGC1α, a transcriptional co-activator of nuclear receptors that is necessary for mitochondrial biogenesis and UCP1 expression.53, 67, 68 Furthermore, FFAs derived from intracellular lipolysis or supplied by circulation are also required for UCP1 activity and serve as a substrate for beta-oxidation and induction of UCP1 expression.7, 69
In addition to neuronal control, hormones, cytokines, and other circulating factors can help control BAT activity. For example, the thyroid hormone triiodothyronine activates BAT function and WAT browning by enhancing mitochondrial biogenesis and UCP1 expression,70, 71 a function that is also supported by in vitro studies on primary mesenchymal stem cell cultures derived from human adipose tissue.72
Various factors have been identified as activators of BAT expansion and beige/brite adipocyte differentiation in WAT. Notably, an anti-diabetic drug, thiazolidinedione, has been shown to increase the metabolic activity of BAT and to be involved in the browning of WAT.33, 73 Moreover, prostaglandins enhance WAT browning, as shown by the pharmacological inhibition and genetic ablation of Cox2.74 Irisin has been identified as a muscle-derived cytokine that is released during exercise and which induces the browning of subcutaneous adipose tissue.75, 76 Bone morphogenetic proteins (BMPs) have been reported to have a critical role not only in early specification of the adipocyte lineage,44 but also in BAT development and function. BMP7 is indispensable for BAT development77 and it can promote beige cell formation and mitochondrial activity in brown adipocytes in vivo and in vitro.78, 79 BMP8b has been found to increase thermogenic activity by acting directly on BAT and by affecting signals derived from the central nervous system.17 Whereas BMP2 and BMP4 are required for white adipogenic commitment of mesenchymal stem cells, an unexpected role of BMP4 in the browning of WAT has been reported.80 Fibroblast growth factor 21 activates BAT activity by elevating PGC1α expression, thereby contributing to the browning of white fat in adults.81, 82 Cardiac natriuretic peptides83 can promote the browning of white fat and thermogenesis in mice and can increase respiration in cultured human fat cells84 through p38 MAP kinase downstream signaling. Dietary factors can also contribute to the control of BAT function. For example, the elevation in FFA levels resulting from a high fat diet increases PPAR activity and induces UCP1 expression and differentiation of brown adipocytes, indicating mechanisms of diet-induced thermogenesis. Moreover, intracellular lipolysis generates FFAs,85 which function as an energy source and also stimulate the thermogenic program of BAT.69 The whitening of BAT has been found to occur in mice deficient in adipose triglyceride lipase, which supports a key role of lipolysis on BAT development. Conversely, overexpression of adipose triglyceride lipase has been shown to enhance the catabolic phenotype of white fat.86
Concluding remarks: therapeutic implications of bat
Several imaging studies on human subjects have shown that BAT prevalence negatively correlates with a high body mass index and fat content, supporting a role of BAT in preventing obesity.87, 88 Moreover, metabolic activity/BAT mass has been reported to be lower in diabetic patients compared with that in non-diabetics.89 Recent studies have assessed oxidative metabolism by using 11C-acetate and have demonstrated clearly that human BAT critically contributes to energy expenditure through cold-induced non-shivering thermogenesis.90 However, the metabolic significance of cold-induced activation of human BAT has been questioned by Muzik et al.,91 whose work showed that upon exposure to moderately cool temperatures, BAT activation accounted for only 15–25 kcal per day.
In rodent models, pharmacological induction of BAT function has been shown to be beneficial in counteracting obesity and diabetes. For instance, ADRB3 agonists have been shown to treat obesity and diabetes.11 However, in humans, ADRB3 agonists are ineffective because ADRB3 is expressed at low levels in adipocytes. The use of indirect sympathomimetics (for example, ephedrine and sibutramine) produces adverse effects such as increased cardiovascular complications and stroke events, which is associated with the broad and non-specific action of adrenergic stimulation. Furthermore, indirect sympathomimetics do not activate human BAT. Therefore, approaches other than β-adrenergic control are required for developing practical applications of pharmacological BAT activation in humans. In this regard, non-canonical thermogenic stimulators, which work independently of adrenergic receptors, could help increase BAT activity without causing adverse outcomes or patient discomfort. Irisin has been identified as a muscle-derived cytokine (myokine) that induces the browning of adipose tissue and BAT activation. Another promising agent is fibroblast growth factor 21. Recent clinical studies have shown that the fibroblast growth factor 21 analog LY2405319 exhibits beneficial effects in patients suffering from obesity and type 2 diabetics by improving their metabolic profiles.92 BAT transplantation or cell-based therapies are also attractive approaches that could be used to expand BAT in vivo. Reversal of type 1 diabetes by means of BAT transplantation has been reported in studies using rodent models.13 Human adipose-derived stem cells and inducible pluripotent stem cells have been used to investigate and understand the signaling that determines progenitor fate and activity during BAT expansion and WAT browning.93, 94 Parameters of the tissue extracellular environment such as vascularization, angiogenesis and levels of innervation should be considered when deciphering the integration of cues that direct context-dependent fine-tuning of progenitor cell behavior.
Although the complexity of the physiological processes required for energy homeostasis has hampered the development of drugs that can effectively treat metabolic diseases, BAT has been proven to function as a crucial regulator in energy metabolism. Considering the substantial clinical benefits of therapeutically manipulating BAT activity and mass, understanding the mechanisms controlling this process will facilitate the development of safe and effective therapeutic tools to combat obesity and metabolic disorders.
References
Haslam D. W., James W. P . Obesity. Lancet 2005; 366: 1197–1209.
Jia H., Lubetkin E. I . Trends in quality-adjusted life-years lost contributed by smoking and obesity. Am. J. Prev. Med. 2010; 38: 138–144.
Whittle A. J., López M., Vidal-Puig A . Using brown adipose tissue to treat obesity – the central issue. Trends Mol. Med. 2011; 17: 405–411.
Galic S., Oakhill J. S., Steinberg G. R . Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010; 316: 129–139.
Glass C. K., Olefsky J. M . Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012; 15: 635–645.
Gesta S., Tseng Y. H., Kahn C. R . Developmental origin of fat: tracking obesity to its source. Cell 2007; 131: 242–256.
Cannon B., Nedergaard J . Brown adipose tissue: function and physiological significance. Physiol. Rev. 2004; 84: 277–359.
Van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D. et al. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009; 360: 1500–1508.
Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T. et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009; 360: 1518–1525.
Granneman J. G., Burnazi M., Zhu Z., Schwamb L. A . White adipose tissue contributes to UCP1-independent thermogenesis. Am. J. Physiol. Endocrinol. Metab. 2003; 285: E1230–E1236.
Ghorbani M., Claus T. H., Himms-Hagen J . Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a beta3-adrenoceptor agonist. Biochem. Pharmacol. 1997; 54: 121–131.
Tseng Y. H., Cypess A. M., Kahn C. R . Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 2010; 9: 465–482.
Gunawardana S. C., Piston D. W . Reversal of type 1 diabetes in mice by brown adipose tissue transplant. Diabetes 2012; 61: 674–682.
Hamann A., Flier J. S., Lowell B. B . Decreased brown fat markedly enhances susceptibility to diet-induced obesity, diabetes, and hyperlipidemia. Endocrinology 1996; 137: 21–29.
Lidell M. E., Enerback S . Brown adipose tissue - a new role in humans? Nat. Rev. Endocrinol. 2010; 6: 319–325.
Bartelt A., Bruns O. T., Reimer R., Hohenberg H., Ittrich H., Peldschus K. et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011; 17: 200–205.
Whittle A., Carobbio S., Martins L., Slawik M., Hondares E., Vázquez M. et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 2012; 149: 871–885.
Arch J. R . β3-Adrenoceptor agonists: potential, pitfalls and progress. Eur. J. Pharmacol. 2002; 440: 99–107.
Zeve D., Tang W., Graff J . Fighting fat with fat: the expanding field of adipose stem cells. Cell Stem Cell 2009; 5: 472–481.
Wu J., Cohen P., Spiegelman B. M . Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013; 27: 234–250.
Sacks H., Symonds M. E . Anatomical locations of human brown adipose tissue: functional relevance and implications in obesity and type 2 diabetes. Diabetes 2013; 62: 1783–1790.
Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S. et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008; 454: 961–967.
Gensch N., Borchardt T., Schneider A., Riethmacher D., Braun T . Different autonomous myogenic cell populations revealed by ablation of Myf5-expressing cells during mouse embryogenesis. Development 2008; 135: 1597–1604.
Sanchez-Gurmaches J., Hung C. M., Sparks C. A., Tang Y., Li H., Guertin D. A . PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab. 2012; 16: 348–362.
Atit R., Sgaier S. K., Mohamed O. A., Taketo M. M., Dufort D., Joyner A. L. et al. β-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev. Biol. 2006; 296: 164–176.
Padilla J., Jenkins N. T., Vieira-Potter V. J., Laughlin M. H . Divergent phenotype of rat thoracic and abdominal perivascular adipose tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013; 304: R543–R552.
Olson L. E., Soriano P . PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev. Cell 2011; 20: 815–826.
Chang L., Villacorta L., Li R., Hamblin M., Xu W., Dou C. et al. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-γ deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 2012; 126: 1067–1078.
Huttunen P., Hirvonen J., Kinnula V . The occurrence of brown adipose tissue in outdoor workers. Eur. J. Appl. Physiol. Occup. Physiol. 1981; 46: 339–345.
Granneman J. G., Li P., Zhu Z., Lu Y . Metabolic and cellular plasticity in white adipose tissue I: effects of β3-adrenergic receptor activation. Am. J. Physiol. Endocrinol. Metab. 2005; 289: E608–E616.
Enerback S . Adipose tissue metabolism in 2012: adipose tissue plasticity and new therapeutic targets. Nat. Rev. Endocrinol. 2013; 9: 69–70.
Wu J., Boström P., Sparks L. M., Ye L., Choi J. H., Giang A. H. et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012; 150: 366–376.
Petrovic N., Walden T. B., Shabalina I. G., Timmons J. A., Cannon B., Nedergaard J . Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010; 285: 7153–7164.
Rosenwald M., Perdikari A., Rülicke T., Wolfrum C . Bi-directional interconversion of brite and white adipocytes. Nat. Cell Biol. 2013; 15: 659–667.
Barbatelli G., Murano I., Madsen L., Hao Q., Jimenez M., Kristiansen K. et al. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am. J. Physiol. Endocrinol. Metab. 2010; 298: E1244–E1253.
Cinti S . Transdifferentiation properties of adipocytes in the adipose organ. Am. J. Physiol. Endocrinol. Metab. 2009; 297: E977–E986.
Himms-Hagen J., Melnyk A., Zingaretti M. C., Ceresi E., Barbatelli G., Cinti S . Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am. J. Physiol. Cell Physiol. 2000; 279: C670–C681.
Lee Y. H., Petkova A. P., Mottillo E. P., Granneman J. G . In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012; 15: 480–491.
Joe A. W., Yi L., Natarajan A., Le Grand F., So L., Wang J. et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010; 12: 153–163.
Uezumi A., Fukada S. I., Yamamoto N., Takeda S. I., Tsuchida K . Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010; 12: 143–152.
Lee Y. H., Petkova A. P., Granneman J. G . Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013; 18: 355–367.
Lidell M. E., Betz M. J., Leinhard O. D., Heglind M., Elander L., Slawik M. et al. Evidence for two types of brown adipose tissue in humans. Nat. Med. 2013; 19: 631–634.
Cypess A. M., White A. P., Vernochet C., Schulz T. J., Xue R., Sass C. A. et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013; 19: 635–639.
Tang Q. Q., Lane M. D . Adipogenesis: from stem cell to adipocyte. Annu. Rev. Biochem. 2012; 81: 715–736.
Cawthorn W. P., Scheller E. L., Macdougald O. A . Adipose tissue stem cells meet preadipocyte commitment: going back to the future. J. Lipid Res. 2012; 53: 227–246.
Harms M., Seale P . Brown and beige fat: development, function and therapeutic potential. Nat. Med. 2013; 19: 1252–1263.
Floyd Z. E., Stephens J. M . Controlling a master switch of adipocyte development and insulin sensitivity: covalent modifications of PPARγ. Biochim. Biophys. Acta 2012; 1822: 1090–1095.
Qiang L., Wang L., Kon N., Zhao W., Lee S., Zhang Y. et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of PPARgamma. Cell 2012; 150: 620–632.
Villanueva C. J., Waki H., Godio C., Nielsen R., Chou W. L., Vargas L. et al. TLE3 is a dual-function transcriptional coregulator of adipogenesis. Cell Metab. 2011; 13: 413–427.
Rajakumari S., Wu J., Ishibashi J., Lim H. W., Giang A. H., Won K. J. et al. EBF2 determines and maintains brown adipocyte identity. Cell Metab. 2013; 17: 562–574.
Sugii S., Evans R. M . Epigenetic codes of PPARγ in metabolic disease. FEBS Lett. 2011; 585: 2121–2128.
Siersbæk M. S., Loft A., Aagaard M. M., Nielsen R., Schmidt S. F., Petrovic N. et al. Genome-wide profiling of peroxisome proliferator-activated receptor γ in primary epididymal, inguinal, and brown adipocytes reveals depot-selective binding correlated with gene expression. Mol. Cell Biol. 2012; 32: 3452–3463.
Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M . A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998; 92: 829–839.
Cao W., Daniel K. W., Robidoux J., Puigserver P., Medvedev A. V., Bai X. et al. p38 Mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 2004; 24: 3057–3067.
Seale P . Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007; 6: 38–54.
Seale P., Conroe H. M., Estall J., Kajimura S., Frontini A., Ishibashi J. et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Inves. 2011; 121: 96–105.
Kajimura S . Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 2009; 460: 1154–1158.
Kajimura S . Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008; 22: 1397–1409.
Ohno H., Shinoda K., Ohyama K., Sharp L. Z., Kajimura S . EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 2013; 504: 163–167.
Villanueva C. J., Vergnes L., Wang J., Drew B. G., Hong C., Tu Y. et al. Adipose subtype-selective recruitment of TLE3 or Prdm16 by PPARγ specifies lipid storage versus thermogenic gene programs. Cell Metab. 2013; 17: 423–435.
Trajkovski M., Ahmed K., Esau C. C., Stoffel M . MyomiR-133 regulates brown fat differentiation through Prdm16. Nat. Cell Biol. 2012; 14: 1330–1335.
Yin H., Pasut A., Soleimani V. D., Bentzinger C. F., Antoun G., Thorn S. et al. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab. 2013; 17: 210–224.
Liu W., Bi P., Shan T., Yang X., Yin H., Wang Y. X. et al. miR-133a regulates adipocyte browning in vivo. PLoS Genet. 2013; 9: e1003626.
Young J. B., Saville E., Rothwell N. J., Stock M. J., Landsberg L . Effect of diet and cold exposure on norepinephrine turnover in brown adipose tissue of the rat. J. Clin. Invest. 1982; 69: 1061–1071.
Murano I., Barbatelli G., Giordano A., Cinti S . Noradrenergic parenchymal nerve fiber branching after cold acclimatisation correlates with brown adipocyte density in mouse adipose organ. J. Anat. 2009; 214: 171–178.
Giordano A., Frontini A., Murano I., Tonello C., Marino M. A., Carruba M. O. et al. Regional-dependent increase of sympathetic innervation in rat white adipose tissue during prolonged fasting. J. Histochem. Cytochem. 2005; 53: 679–687.
Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B. M . Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006; 3: 333–341.
Chang J. S., Fernand V., Zhang Y., Shin J., Jun H. J., Joshi Y. et al. NT-PGC-1alpha protein is sufficient to link beta3-adrenergic receptor activation to transcriptional and physiological components of adaptive thermogenesis. J. Biol. Chem. 2012; 287: 9100–9111.
Mottillo E. P., Bloch A. E., Leff T., Granneman J. G . Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 2012; 287: 25038–25048.
Villarroya F., Vidal-Puig A . Beyond the sympathetic tone: the new brown fat activators. Cell Metab. 2013; 17: 638–643.
Lopez M., Varela L., Vazquez M. J., Rodriguez-Cuenca S., Gonzalez C. R., Velagapudi V. R. et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010; 16: 1001–1008.
Lee J. Y., Takahashi N., Yasubuchi M., Kim Y. I., Hashizaki H., Kim M. J. et al. Triiodothyronine induces UCP-1 expression and mitochondrial biogenesis in human adipocytes. Am. J. Physiol. Cell Physiol. 2012; 302: C463–C472.
Wilson-Fritch L., Burkart A., Bell G., Mendelson K., Leszyk J., Nicoloro S. et al. Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol. Cell. Biol. 2003; 23: 1085–1094.
Vegiopoulos A., Muller-Decker K., Strzoda D., Schmitt I., Chichelnitskiy E., Ostertag A. et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010; 328: 1158–1161.
Boström P., Wu J., Jedrychowski M. P., Korde A., Ye L., Lo J. C. et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012; 481: 463–468.
Raschke S., Elsen M., Gassenhuber H., Sommerfeld M., Schwahn U., Brockmann B. et al. Evidence against a beneficial effect of irisin in humans. PLoS One 2013; 8: e73680.
Tseng Y. H . New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008; 454: 1000–1004.
Schulz T. J., Huang T. L., Tran T. T., Zhang H., Townsend K. L., Shadrach J. L. et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc. Natl Acad. Sci. USA 2011; 108: 143–148.
Townsend K. L., An D., Lynes M. D., Huang T. L., Zhang H., Goodyear L. J. et al. Increased mitochondrial activity in BMP7-treated brown adipocytes, due to increased CPT1- and CD36-mediated fatty acid uptake. Antioxid. Redox Signal 2013; 19: 243–257.
Qian S. W., Tang Y., Li X., Liu Y., Zhang Y. Y., Huang H. Y. et al. BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc. Natl Acad. Sci. 2013; 110: E798–E807.
Fisher F., Kleiner S., Douris N., Fox E., Mepani R., Verdeguer F. et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012; 26: 271–281.
Chartoumpekis D., Habeos I., Ziros P., Psyrogiannis A., Kyriazopoulou V., Papavassiliou A . Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol. Med. 2011; 17: 736–740.
Lafontan M., Moro C., Berlan M., Crampes F., Sengenes C., Galitzky J . Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol. Metab. 2008; 19: 130–137.
Bordicchia M., Liu D., Amri E. Z., Ailhaud G., Dessì-Fulgheri P., Zhang C. et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest. 2012; 122: 1022–1036.
Moore H. P. H., Silver R. B., Mottillo E. P., Bernlohr D. A., Granneman J. G . Perilipin targets a novel pool of lipid droplets for lipolytic attack by hormone-sensitive lipase. J. Biol. Chem. 2005; 280: 43109–43120.
Ahmadian M., Abbott M. J., Tang T., Hudak C. S., Kim Y., Bruss M. et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011; 13: 739–748.
Frontini A., Cinti S . Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 2010; 11: 253–256.
Saito M., Okamatsu-Ogura Y., Matsushita M., Watanabe K., Yoneshiro T., Nio-Kobayashi J. et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 2009; 58: 1526–1531.
Ouellet V., Routhier-Labadie A., Bellemare W., Lakhal-Chaieb L., Turcotte E., Carpentier A. C. et al. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J. Clin. Endocrinol. Metab. 2011; 96: 192–199.
Ouellet V., Labbé S. M., Blondin D. P., Phoenix S., Guérin B., Haman F. et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Invest. 2012; 122: 545–552.
Muzik O., Mangner T. J., Leonard W. R., Kumar A., Janisse J., Granneman J. G . 15O PET measurement of blood flow and oxygen consumption in cold-activated human brown fat. J. Nucl. Med. 2013; 54: 523–531.
Gaich G., Chien J. Y., Fu H., Glass L. C., Deeg M. A., Holland W. L. et al. The Effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013; 18: 333–340.
Ahfeldt T., Schinzel R. T., Lee Y. K., Hendrickson D., Kaplan A., Lum D. H. et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012; 14: 209–219.
Cawthorn W. P., Scheller E. L., Macdougald O. A . Adipose tissue stem cells: the great WAT hope. Trends Endocrinol. Metab. 2012; 23: 270–277.
Lee Y. H., Mottillo E. P., Granneman J. G . Adipose tissue plasticity from WAT to BAT and in between. Biochim. Biophys. Acta. 2013; 13: 165–168.
Acknowledgements
We would like to thank Dr James G Granneman for valuable advice and insightful comments on the manuscript. This research was partly supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0023938).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lee, YH., Jung, YS. & Choi, D. Recent advance in brown adipose physiology and its therapeutic potential. Exp Mol Med 46, e78 (2014). https://doi.org/10.1038/emm.2013.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.163
Keywords
This article is cited by
-
Ssu72 phosphatase is essential for thermogenic adaptation by regulating cytosolic translation
Nature Communications (2023)
-
Brown adipose tissue and alzheimer’s disease
Metabolic Brain Disease (2023)
-
Quercetin, Engelitin and Caffeic Acid of Smilax china L. Polyphenols, Stimulate 3T3-L1 Adipocytes to Brown-like Adipocytes Via β3-AR/AMPK Signaling Pathway
Plant Foods for Human Nutrition (2022)
-
Development of CIDEA reporter mouse model and its application for screening thermogenic drugs
Scientific Reports (2021)
-
Inflammation and tumor progression: signaling pathways and targeted intervention
Signal Transduction and Targeted Therapy (2021)
