





Abstract
The N-methyl-d-aspartate (NMDA) receptor antagonist ketamine has rapid antidepressant effects in treatment-resistant major depressive disorder (MDD). In rats, ketamine selectively increased electroencephalogram (EEG) slow wave activity (SWA) during non-rapid eye movement (REM) sleep and altered central brain-derived neurotrophic factor (BDNF) expression. Taken together, these findings suggest that higher SWA and BDNF levels may respectively represent electrophysiological and molecular correlates of mood improvement following ketamine treatment. This study investigated the acute effects of a single ketamine infusion on depressive symptoms, EEG SWA, individual slow wave parameters (surrogate markers of central synaptic plasticity) and plasma BDNF (a peripheral marker of plasticity) in 30 patients with treatment-resistant MDD. Montgomery–Åsberg Depression Rating Scale scores rapidly decreased following ketamine. Compared to baseline, BDNF levels and early sleep SWA (during the first non-REM episode) increased after ketamine. The occurrence of high amplitude waves increased during early sleep, accompanied by an increase in slow wave slope, consistent with increased synaptic strength. Changes in BDNF levels were proportional to changes in EEG parameters. Intriguingly, this link was present only in patients who responded to ketamine treatment, suggesting that enhanced synaptic plasticity – as reflected by increased SWA, individual slow wave parameters and plasma BDNF – is part of the physiological mechanism underlying the rapid antidepressant effects of NMDA antagonists. Further studies are required to confirm the link found here between behavioural and synaptic changes, as well as to test the reliability of these central and peripheral biomarkers of rapid antidepressant response.
Introduction
Currently available antidepressants, which primarily target the noradrenergic and serotonergic systems, often successfully alleviate depressive symptoms in individuals with major depressive disorder (MDD); however, they are associated with several limitations, including delayed therapeutic onset. Thus, the development of novel agents characterized by more rapid onset of antidepressant effects would have a substantial impact on both patient care and public health. In this regard, recent studies employing glutamate-modulating compounds to treat MDD have demonstrated the rapid onset of antidepressant effects of these drugs (aan het Rot et al.2010; Berman et al.2000; Diazgranados et al.2010; Maeng & Zarate, 2007; Preskorn et al.2008; Valentine et al.2011; Zarate et al.2004, 2006a, b). More specifically, recent studies have demonstrated that a single infusion of ketamine hydrochloride rapidly and significantly improved depressive symptoms within 2 h. Extensive research into the neuronal and molecular mechanisms underlying the rapid antidepressant effects of N-methyl-d-aspartate (NMDA) antagonists such as ketamine is ongoing. Preclinical studies found that ketamine injections increased synaptic strength in rat prefrontal cortex, as reflected by: (a) enhanced post-synaptic protein signalling; (b) increased synaptogenesis; (c) enhanced excitatory post-synaptic currents (Li et al.2010).
Electroencephalographic (EEG) studies in rats showed that peritoneal injections of ketamine (Feinberg & Campbell, 1993) and dizocilpine-maleate (MK-801) – both of which are NMDA antagonists (Campbell & Feinberg, 1996) – increased sleep slow wave activity (SWA; EEG activity between 1 and 4 Hz) during non-rapid eye movement (NREM) sleep. A relationship between SWA and cortical synaptic activity of cortical neurons was later demonstrated in an extensive set of experiments employing high-density EEG in humans, local field potentials in rats and large-scale simulations. These experiments established that SWA and individual slow wave parameters (incidence, amplitude and slope) are sensitive markers of cortical synaptic strength and network synchronization (Esser et al.2007; Riedner et al.2007; Vyazovskiy et al.2007).
At the molecular level, brain-derived neurotrophic factor (BDNF) has a key role in synaptic potentiation (Lu, 2003) and long-term potentiation (Kossel et al.2001; Messaoudi et al.2002, 2007) and is a major contributor to neuronal plasticity. Furthermore, a recent study found that cortical unilateral microinjections of BDNF led to regionally specific increases in SWA (Faraguna et al.2008). With regard to MDD, lower levels of serum BDNF have been found in antidepressant-naive patients with MDD compared to those receiving antidepressants or to normal controls. In addition, BDNF levels were higher in patients who responded to antidepressant drugs than those who did not (Karege et al.2002a; Sen et al.2008) and were also higher in those patients responding to sleep deprivation (Gorgulu & Caliyurt, 2009). Another recent study noted a causal association between ketamine's antidepressant-like effects and increased activity-dependent BDNF in rodents (Autry et al.2011).
This study sought to investigate whether ketamine increases synaptic strength in 30 drug-free treatment-resistant MDD patients, using SWA and slow wave parameters, as well as plasma BDNF, as central and peripheral biomarkers of neuroplasticity. Specifically, we assessed the acute effects of a single infusion of ketamine on NREM sleep EEG, plasma BDNF and depressive symptoms assessed using the Montgomery–Åsberg Rating Scale (MADRS; Montgomery & Asberg, 1979). We predicted that mood ratings would improve and that SWA, plasma BDNF levels and the incidence, amplitude and slope of slow waves would proportionally increase during mood improvement after ketamine infusion.
Method
MDD patients
Thirty patients (20 male, 10 female; mean age 48.06 ± 2.34 yr) with a current diagnosis of MDD without psychotic features were studied at the National Institute of Mental Health Clinical Research Center Mood Disorders Research Unit in Bethesda, Maryland. Diagnosis was confirmed using the Structured Clinical Interview for Axis I DSM-IV Disorders-Patient version. Inclusion criteria were a MADRS score ⩾22 and a current or past history of non-response to two adequate antidepressant trials. Exclusion criteria included a DSM-IV diagnosis of drug or alcohol dependence or abuse in the last 3 months, serious, unstable medical illness (including neurological disease and head trauma) or uncorrected hypo- or hyperthyroidism. Anxiety disorders were not exclusionary. All patients were free from any psychotropic drug for at least 2 wk (5 wk for fluoxetine) before ketamine infusion. Cigarette use was permitted during the clinical trial, but alcohol use was not.
The mean MADRS score for the patient group was 32.5 ± 0.85. Depressive symptoms were examined via MADRS ratings conducted at baseline (60 min before ketamine infusion), at 230 min post-infusion, 1 d post-infusion and 2 d post-infusion. At all time-points, change in depressive symptoms was expressed as change in score from baseline, with negative values reflecting a reduction in symptoms.
The study was approved by the Combined Neuroscience Institutional Review Board of the National Institutes of Health. All subjects provided written informed consent before entry into the study.
Drug administration
Ketamine infusion was conducted as previously described (Zarate et al.2006a). Briefly, at about 10:00 hours, MDD patients openly received a single i.v. infusion of 0.5 mg/kg ketamine hydrochloride over the course of 40 min. The present study was designed to examine electrophysiological and molecular correlates as biomarkers of acute response (first day) to ketamine in patients with treatment-resistant MDD. The study was part of a larger protocol investigating the maintenance of clinical effects in these patients using riluzole, another glutamatergic modulator (Ibrahim et al.2012). The present sleep EEG substudy was begun after about one-third of the subjects had entered the study by Ibrahim and colleagues, thus resulting in a different total number of subjects. However, other than the one subject excluded due to technical EEG problems, all subsequent subjects enrolled in the larger study were included in the present study. Therefore, 4–6 h after ketamine infusion, patients were randomized to receive either double-blind riluzole [N = 19; 100 mg/d; 50 mg b.i.d. (twice per day)] or placebo (N = 11). Raters were blind to riluzole treatment. Riluzole has multiple effects on the glutamate system. However, previous studies have noted that its therapeutic onset is delayed (Zarate et al.2004); thus, we did not expect to find differences in the acute phase between the different treatment groups. Nevertheless, because an early interactive effect between the two compounds could not be excluded, we controlled for this possibility by analysing group differences before merging the two groups.
EEG recordings
Following an adaptation night, whole night sleep recordings were obtained for all 30 patients on the night before ketamine infusion as well as on the two following nights. EEG recordings were performed approximately 12 h after the ketamine infusion in order to prevent potential residual neurotoxic effects (Campbell & Feinberg, 1996). Two EEGs (C3/A2 and C4/A1), two electrooculograms and one submental electromyogram were recorded using a Nihon–Kohden system (Neurofax Sleep v. 05-50; Nihon Kohden Corporation, Japan) and Polysmith Acquisition and Review software (v. 4.0.25.0; Nihon Kohden) with a 200 Hz sampling rate. One patient was excluded due to technical EEG problems. Sleep EEGs were visually scored by blind reviewers in 30-s epochs according to established criteria (Rechtschaffen & Kales, 1968).
EEG spectral analysis
Signals from C3/A2 and C4/A1 derivations were filtered (0.5–30 Hz) and spectral density analysis was conducted using 0.2 Hz bin resolution (Welch's averaged modified periodogram with a Hamming window, averages of six 5-s epochs). NREM sleep epochs (30 s) that exceeded a threshold (eight times the mean power values) in the 0.6–4.6 and 20–30 Hz frequency bands were excluded from the analysis (<10% for each night). SWA was calculated as the average power spectral density between 0.6 and 4 Hz for each EEG spectral derivation during NREM sleep. Because SWA values were virtually identical at C3 and C4, averaged SWA values are presented in the Results section.
Analysis of slow wave parameters
Slow wave parameters were calculated by applying a recently developed procedure adopting fixed parameters derived from sleep EEG standard guidelines (Riedner et al.2007). In order to detect individual slow waves, EEG signals were down-sampled to 100 Hz and band-passed (0.5–4 Hz, stop-band at 0.1 and 10 Hz) using a Chebyshev Type II filter (MATLAB; MathWorks Inc., USA). Waves were detected as negative deflections between two consecutive zero crossings. Negative deflections were chosen because of their stability compared to the high degree of variability associated with positive deflections. Only waves with 0.25- to 1.0-s consecutive zero crossings detected in artefact-free NREM epochs were considered slow waves. Wave number and negative peak amplitude distribution were calculated for each EEG derivation. As with SWA analysis, the presented slow wave parameters were averaged across the two EEG derivations.
BDNF analysis
BDNF was collected using the vacutainer system before ketamine infusion (∼09:00 hours), as well as 230 min after ketamine infusion (∼14:00 hours), before riluzole/placebo was administered. A blood sample could not be collected from one subject (n = 29). Blood samples were centrifuged at 216 g at 4 °C for 5 min and stored at −80 °C until assay. When ready for assay, an anti-BDNF sandwich ELISA kit (Chemicon International, USA) was used according to the manufacturer's instructions in order to measure BDNF. The assay was performed blind to clinical information. The plasma was diluted 1:2 with a sample buffer and the assay performed in duplicate. In order to create the standard curve for BDNF levels, a BDNF standard solution was diluted from 7.8 to 500 pg/ml concentrations in a microplate reader. Streptavidin enzyme, substrate and stop solution were added and BDNF levels were then determined by absorbance in 450 nm using optical density values based on the standard curve values.
Statistical analyses
Separate linear mixed models were used for EEG and MADRS scores. For treatment comparisons, the between-subject factor drug (ketamine/placebo; ketamine/riluzole) was used to neutralize potential confounds due to the different treatments. If not normally distributed, data were log-transformed prior to further analysis. Changes in MADRS scores were investigated via the within-subject factor time (1 h before infusion; 230 min post-infusion; 1 d post-infusion; 2 d post-infusion), which was used to assess the time-course of the effects of ketamine infusion on mood. For SWA and slow wave parameters, the within-subject factor night (baseline; day of infusion; second night) was used to determine the time-course of the effects of the infusion on sleep EEG. Significant effects were examined with simple effects tests; Bonferroni's correction for multiple comparisons was used where appropriate. BDNF differences were examined using a paired t test. Significance was evaluated at p < 0.05, two-tailed.
Results
Ketamine infusion reduced depressive symptoms
After treatment with ketamine/placebo or ketamine/riluzole, patients’ depressive symptoms – as assessed by the MADRS – were significantly improved. As reported previously (Ibrahim et al.2012), no significant effect was noted for drug, either as a main effect (F1,28 = 0.006; p = 0.93) or in an interaction with time (F3,84 = 0.43; p = 0.72), suggesting that the two treatment conditions (ketamine/placebo and ketamine/riluzole) had similar effects. When combining the two groups, depressive symptoms were found to be improved for all the observed time-points (p < 0.01; Fig. 1a). No serious adverse events occurred during the study (Ibrahim et al.2012) nor were differences in minor adverse reactions observed between patients who did and did not respond to ketamine. Mood elevation assessed by Young Mania Rating Scale (YMRS) scores did occur in six subjects (14.2%); three of these met response criteria. No correlation was observed between YMRS scores and decrease in MADRS scores.
![(a) Montgomery–Asberg Depression Rating Scale (MADRS) scores before (−60 min) and after ketamine infusion (230 min, day 1, day 2). Mixed analysis of variance (F3,84 = 29.48; p < 0.00001) and simple effects tests (Bonferroni's corrected) indicated that, compared to pre-infusion baseline, mood was significantly improved at 230 min post-infusion (41.46 ± 6.62%; p < 0.00001), at 1 d post-infusion (40.38 ± 6.61%; p < 0.00001) and at 2 d post-infusion (39.75 ± 6.54%; p < 0.00001). (b) Plasma log brain-derived neurotrophic factor (BDNF) levels measured before (−60 min) and after (230 min) ketamine infusion [actual means at −60 and 230 were 1948.1 ± 402.2 (s.e.m.) and 2303.7 ± 387.2 (s.e.m.), respectively]. The boxes indicate the 25th/75th percentile; the dashed line within the boxes marks the median. Whiskers above and below the box indicate the 90th and 10th percentiles, respectively. Empty dots represent outliers.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ijnp/16/2/10.1017_S1461145712000545/1/m_s1461145712000545_fig1.gif?Expires=1716334180&Signature=z2QgP9zcJgmsW5zVbaD19GgVvKziP4Rn4Jq3eLatU6Ltd7OZD6~NXi5w2lHCiT1OOibjgU~qyGTO7aa-rJmOLdxFTxlt~nqoLDfnYAe~wzClLdGabnLjR8MnP1JUlLhKeUQsJASvpAzYeb1Ixstr7-VfvrMVA3AxHc-m2NSt4GPkJ5wsZXKvb4DGuHYMhbGwOY7uu7NtCXX7VVGWf77GPH6Qz~lVVdZZE9GqUtqhoRoJq4VxZwYj2Rwnhl2cWpoiRfY3ZcJBzNZeVW44MZcj1wtM1CljWRTyQS~wdqMRqPeijohUO3akhBuHC976DjZ4GZeo5ERBM8E9ythFmnemnw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(a) Montgomery–Asberg Depression Rating Scale (MADRS) scores before (−60 min) and after ketamine infusion (230 min, day 1, day 2). Mixed analysis of variance (F3,84 = 29.48; p < 0.00001) and simple effects tests (Bonferroni's corrected) indicated that, compared to pre-infusion baseline, mood was significantly improved at 230 min post-infusion (41.46 ± 6.62%; p < 0.00001), at 1 d post-infusion (40.38 ± 6.61%; p < 0.00001) and at 2 d post-infusion (39.75 ± 6.54%; p < 0.00001). (b) Plasma log brain-derived neurotrophic factor (BDNF) levels measured before (−60 min) and after (230 min) ketamine infusion [actual means at −60 and 230 were 1948.1 ± 402.2 (s.e.m.) and 2303.7 ± 387.2 (s.e.m.), respectively]. The boxes indicate the 25th/75th percentile; the dashed line within the boxes marks the median. Whiskers above and below the box indicate the 90th and 10th percentiles, respectively. Empty dots represent outliers.
Several patients still showed significant depressive symptoms after ketamine infusion (Fig. 1a). Consistent with our prior work (Zarate et al.2006a), we defined those patients exhibiting a reduction of ⩾50% in MADRS scores at 230 min post-infusion as ketamine responders (n = 13); non-responders (n = 17) were those with <50% improvement.
Ketamine infusion increased log BDNF levels
Ketamine infusion was associated with significantly increased BDNF plasma levels at 230 min post-infusion relative to the baseline measurement [t = 2.116, p < 0.05; degrees of freedom (df) = 28; Fig. 1b].
Ketamine infusion affected sleep parameters
A linear mixed model was used to test the effects of drug and night on several sleep parameters; results are listed in Table 1. Again, no significant effects for drug were noted and the analysis of variance (ANOVA) values presented in Table 1 refer to the main effects of night. Compared to the baseline night, total sleep time was significantly increased (p < 0.05) after ketamine infusion, as were slow wave sleep and REM. In addition, S1, S2, REM latency, and waking time on the day after infusion were all reduced (p < 0.05), indicating that sleep was more consolidated after ketamine infusion.
Impact of ketamine infusion on sleep (n = 30)
| Variables | BSL (1) Mean (±s.e.m.) | Infusion (2) Mean (±s.e.m.) | Recovery (3) Mean (±s.e.m.) | ANOVA | p value for comparisona | ||||
|---|---|---|---|---|---|---|---|---|---|
| df | F | p | 1 vs. 2 | 2 vs. 3 | 1 vs. 3 | ||||
| TST, min | 368.10 (10.4) | 392.97 (7.63) | 395.00 (8.35) | 2, 58 | 5.5 | <0.01 | 0.02 | 1.00 | 0.01 |
| S1, % | 9.46 (0.93) | 7.79 (0.71) | 8.84 (0.81) | 2, 58 | 3.3 | <0.05 | 0.03 | 0.33 | 1.00 |
| S2, % | 62.11 (1.80) | 58.32 (2.14) | 59.63 (1.98) | 2, 58 | 4.74 | 0.01 | 0.001 | 0.14 | 0.87 |
| SWS, % | 3.52 (0.70) | 4.73 (0.99) | 4.40 (0.92) | 2, 58 | 16.6 | 0.01 | 0.04 | 1.00 | 0.20 |
| REM, % | 25.28 (1.30) | 29.51 (1.59) | 27.72 (1.28) | 2, 58 | 5.9 | <0.01 | 0.001 | 0.38 | 0.11 |
| SOL, min | 24.11 (3.7) | 15.56 (2.26) | 18.23 (3.1) | 2, 58 | 4.1 | <0.05 | 0.01 | 1.00 | 0.13 |
| REML, min | 78.71 (6.9) | 61.23 (6.6) | 66.71 (6.5) | 2, 58 | 4.6 | 0.01 | 0.01 | 1.00 | 0.13 |
| Waking, % rec | 17.37 (2.16) | 13.48 (1.43) | 13.25 (1.61) | 2, 58 | 3.5 | <0.05 | 0.06 | 1.00 | 0.04 |
| Variables | BSL (1) Mean (±s.e.m.) | Infusion (2) Mean (±s.e.m.) | Recovery (3) Mean (±s.e.m.) | ANOVA | p value for comparisona | ||||
|---|---|---|---|---|---|---|---|---|---|
| df | F | p | 1 vs. 2 | 2 vs. 3 | 1 vs. 3 | ||||
| TST, min | 368.10 (10.4) | 392.97 (7.63) | 395.00 (8.35) | 2, 58 | 5.5 | <0.01 | 0.02 | 1.00 | 0.01 |
| S1, % | 9.46 (0.93) | 7.79 (0.71) | 8.84 (0.81) | 2, 58 | 3.3 | <0.05 | 0.03 | 0.33 | 1.00 |
| S2, % | 62.11 (1.80) | 58.32 (2.14) | 59.63 (1.98) | 2, 58 | 4.74 | 0.01 | 0.001 | 0.14 | 0.87 |
| SWS, % | 3.52 (0.70) | 4.73 (0.99) | 4.40 (0.92) | 2, 58 | 16.6 | 0.01 | 0.04 | 1.00 | 0.20 |
| REM, % | 25.28 (1.30) | 29.51 (1.59) | 27.72 (1.28) | 2, 58 | 5.9 | <0.01 | 0.001 | 0.38 | 0.11 |
| SOL, min | 24.11 (3.7) | 15.56 (2.26) | 18.23 (3.1) | 2, 58 | 4.1 | <0.05 | 0.01 | 1.00 | 0.13 |
| REML, min | 78.71 (6.9) | 61.23 (6.6) | 66.71 (6.5) | 2, 58 | 4.6 | 0.01 | 0.01 | 1.00 | 0.13 |
| Waking, % rec | 17.37 (2.16) | 13.48 (1.43) | 13.25 (1.61) | 2, 58 | 3.5 | <0.05 | 0.06 | 1.00 | 0.04 |
BSL, Baseline; ANOVA, analysis of variance; TST, total sleep time; S1, non-rapid eye movement (NREM) stage 1 (% of TST); S2, NREM stage 2 (% of TST); SWS, slow wave sleep; REM, stage REM (% of TST); SOL, sleep onset latency; REML, REM sleep latency; rec, recording time.
Post-hoc t tests. p values are expressed with Bonferroni's correction where p < 0.05 is considered significant.
Impact of ketamine infusion on sleep (n = 30)
| Variables | BSL (1) Mean (±s.e.m.) | Infusion (2) Mean (±s.e.m.) | Recovery (3) Mean (±s.e.m.) | ANOVA | p value for comparisona | ||||
|---|---|---|---|---|---|---|---|---|---|
| df | F | p | 1 vs. 2 | 2 vs. 3 | 1 vs. 3 | ||||
| TST, min | 368.10 (10.4) | 392.97 (7.63) | 395.00 (8.35) | 2, 58 | 5.5 | <0.01 | 0.02 | 1.00 | 0.01 |
| S1, % | 9.46 (0.93) | 7.79 (0.71) | 8.84 (0.81) | 2, 58 | 3.3 | <0.05 | 0.03 | 0.33 | 1.00 |
| S2, % | 62.11 (1.80) | 58.32 (2.14) | 59.63 (1.98) | 2, 58 | 4.74 | 0.01 | 0.001 | 0.14 | 0.87 |
| SWS, % | 3.52 (0.70) | 4.73 (0.99) | 4.40 (0.92) | 2, 58 | 16.6 | 0.01 | 0.04 | 1.00 | 0.20 |
| REM, % | 25.28 (1.30) | 29.51 (1.59) | 27.72 (1.28) | 2, 58 | 5.9 | <0.01 | 0.001 | 0.38 | 0.11 |
| SOL, min | 24.11 (3.7) | 15.56 (2.26) | 18.23 (3.1) | 2, 58 | 4.1 | <0.05 | 0.01 | 1.00 | 0.13 |
| REML, min | 78.71 (6.9) | 61.23 (6.6) | 66.71 (6.5) | 2, 58 | 4.6 | 0.01 | 0.01 | 1.00 | 0.13 |
| Waking, % rec | 17.37 (2.16) | 13.48 (1.43) | 13.25 (1.61) | 2, 58 | 3.5 | <0.05 | 0.06 | 1.00 | 0.04 |
| Variables | BSL (1) Mean (±s.e.m.) | Infusion (2) Mean (±s.e.m.) | Recovery (3) Mean (±s.e.m.) | ANOVA | p value for comparisona | ||||
|---|---|---|---|---|---|---|---|---|---|
| df | F | p | 1 vs. 2 | 2 vs. 3 | 1 vs. 3 | ||||
| TST, min | 368.10 (10.4) | 392.97 (7.63) | 395.00 (8.35) | 2, 58 | 5.5 | <0.01 | 0.02 | 1.00 | 0.01 |
| S1, % | 9.46 (0.93) | 7.79 (0.71) | 8.84 (0.81) | 2, 58 | 3.3 | <0.05 | 0.03 | 0.33 | 1.00 |
| S2, % | 62.11 (1.80) | 58.32 (2.14) | 59.63 (1.98) | 2, 58 | 4.74 | 0.01 | 0.001 | 0.14 | 0.87 |
| SWS, % | 3.52 (0.70) | 4.73 (0.99) | 4.40 (0.92) | 2, 58 | 16.6 | 0.01 | 0.04 | 1.00 | 0.20 |
| REM, % | 25.28 (1.30) | 29.51 (1.59) | 27.72 (1.28) | 2, 58 | 5.9 | <0.01 | 0.001 | 0.38 | 0.11 |
| SOL, min | 24.11 (3.7) | 15.56 (2.26) | 18.23 (3.1) | 2, 58 | 4.1 | <0.05 | 0.01 | 1.00 | 0.13 |
| REML, min | 78.71 (6.9) | 61.23 (6.6) | 66.71 (6.5) | 2, 58 | 4.6 | 0.01 | 0.01 | 1.00 | 0.13 |
| Waking, % rec | 17.37 (2.16) | 13.48 (1.43) | 13.25 (1.61) | 2, 58 | 3.5 | <0.05 | 0.06 | 1.00 | 0.04 |
BSL, Baseline; ANOVA, analysis of variance; TST, total sleep time; S1, non-rapid eye movement (NREM) stage 1 (% of TST); S2, NREM stage 2 (% of TST); SWS, slow wave sleep; REM, stage REM (% of TST); SOL, sleep onset latency; REML, REM sleep latency; rec, recording time.
Post-hoc t tests. p values are expressed with Bonferroni's correction where p < 0.05 is considered significant.
Ketamine infusion increased sleep SWA
For both treatment conditions (ketamine/placebo vs. ketamine/riluzole), total SWA amount during the first three NREM episodes of the three recording nights was calculated. These SWA values were normalized for each subject by the SWA of the whole baseline night. The effects of drug, night and sleep cycle were tested using a mixed linear model. Once again, drug had no main effect on SWA (F1,28 = 0.88; p = 0.35), nor was there an interaction between drug and night (F2,56 = 2.35; p = 0.1), sleep cycle (F(2,56) = 0.11; p = 0.88) or night and sleep cycle (F4,112 = 1.89; p = 0.11). When the drug factor was collapsed, SWA was increased on the night of the infusion compared to baseline (p < 0.01). Simple effects tests confirmed that SWA was increased, particularly during the first NREM sleep cycle on the night of ketamine infusion compared to baseline (p < 0.01; Fig. 2a). Furthermore, SWA did not increase within waking EEG epochs prior to sleep onset, indicating that SWA increases were sleep-specific (data not shown).
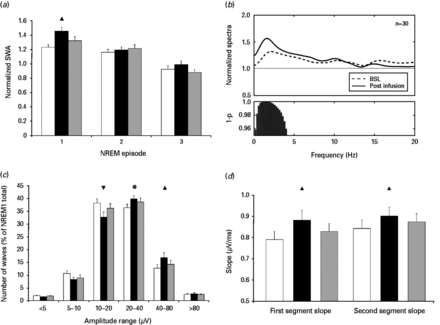
(a) Slow wave activity (SWA) in the first three non-rapid eye movement (NREM) sleep episodes. Mixed analysis of variance (ANOVA) revealed a significant main effect for night (F2,56 = 7.55; p < 0.01). Simple effects tests (Bonferroni's corrected) indicated that SWA was significantly increased on the night of the infusion compared to baseline (p < 0.01). Sleep cycle also significantly affected SWA (F2,56 = 39.9; p < 0.0001). Simple effects tests (Bonferroni's corrected) showed a progressive decline of SWA across the sleep cycles (cycle 1 vs. cycle 2: p < 0.001; cycle 2 vs. cycle 3: p < 0.00001; cycle 1 vs. cycle 3: p < 0.0000001). Finally, a significant interaction was seen between night and sleep cycle (F4,112 = 2.81; p < 0.05), demonstrating that SWA was increased during the first NREM sleep cycle on the night of ketamine infusion compared to baseline. (b) Normalized average power spectra for the first NREM episode of the baseline night (dashed line) and the night after ketamine infusion (solid line). Power values for each frequency bin have been normalized by the average power value of the corresponding bin across the whole baseline night NREM. Bars at the bottom represent significantly different bins (p < 0.05, uncorrected). (c) Distribution of slow wave negative peak amplitude during the first NREM episode. Detected waves over the course of three nights were assigned to logarithmically increasing amplitude ranges based on their negative peak amplitude. Mixed ANOVA (F10,280 = 6.35; p < 0.00001) and simple effects tests (Bonferroni's corrected) demonstrated that, after the ketamine infusion, an overall increase in slow wave amplitude was observed, with a concomitant reduction in low-amplitude waves (10–20 µV negative peak) and increase in higher-amplitude waves (40–80 µV negative peak). (d) First and second-segment average slope of slow waves during the first NREM episode. Mixed ANOVA (F2,56 = 6.7569; p < 0.01 and F2,56 = 5.6104, p < 0.01, respectively) and simple effects tests (Bonferroni's corrected) showed an increase of both segment slopes after ketamine infusion. Triangles indicate significance and direction of the simple effects test (p < 0.01). * Indicate a trend toward significance (p < 0.1). Baseline night is indicated by white bars (a, c and d). The night after the infusion (night 2) is indicated by black bars (a, c and d). Night 3 is indicated by grey bars (a, c and d).
The effects of ketamine infusion on sleep EEG were specific to low frequencies
We further examined other frequency ranges to assess the specificity of ketamine's effects. Relative to the baseline first NREM sleep episode, ketamine infusion significantly increased EEG frequencies restricted to the SWA range (max significant bin: 4 Hz) (Fig. 2b).
Ketamine infusion increased the mean amplitude and slope of slow waves
In order to better characterize the effects of ketamine infusion on synaptic strength and cortical synchronization, we performed separate analyses for total number, amplitude distribution, slope and incidence (number of waves per minute) of automatically detected slow waves during the first NREM sleep episode. For each parameter, separate linear mixed models, including the effects of drug and night, were applied. Drug had no significant effect on any of the slow wave parameters investigated (data not shown); consistent with the SWA findings, treatment groups were therefore combined for subsequent analyses.
Although the total number of slow waves was not affected by ketamine infusion (F2,56 = 0.61; p = 0.54), amplitude distribution analysis showed a marked redistribution of the amplitude of slow waves toward high amplitude (p < 0.01; Fig. 2c). Average slope, calculated on all detected events as the amplitude of the most negative peak divided by the time from the previous zero crossing (first-segment average slope) or the time until the next zero crossing (second-segment average slope), was analysed separately and was significantly increased after ketamine infusion (p < 0.01; Fig. 2d).
Changes in log BDNF level from baseline to 230 min were associated with changes in slow waves on the night of ketamine infusion
Next, the relationship between ketamine-induced changes in peripheral BDNF levels with changes in EEG sleep slow waves group data was examined. EEG change scores were calculated for SWA and for the incidence of high amplitude (20–80 µV) sleep slow waves as the difference between the night of the infusion and the baseline night. These parameters were measured during the first NREM period when the strongest effects of ketamine on SWA were observed. A significant positive correlation (r = 0.45, p = 0.014) between changes in log BDNF and changes in relative SWA (Fig. 3a) was seen. A significant positive correlation (r = 0.41, p = 0.026) between changes in log BDNF and changes in the incidence of high amplitude sleep slow waves (Fig. 3a) was also noted. Partial correlations controlling for possible age effects on BDNF showed that age did not contribute to these effects. BDNF levels were not collected on the full patient sample on the next day (recovery day 3), so the relationship between EEG sleep slow wave and BDNF could not be examined at this time-point.
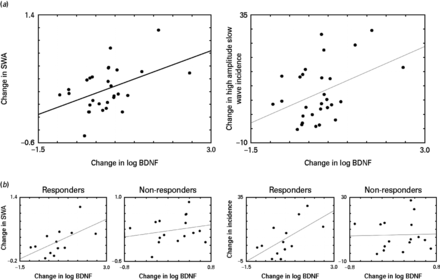
(a, left) The correlation between log brain-derived neurotrophic factor (BDNF) and slow wave activity (SWA) change scores (r = 0.45, p = 0.014). (a, right) The correlation between log BDNF and the incidence of high amplitude wave change scores (r = 0.41, p = 0.026). SWA and high amplitude slow waves incidence change scores were calculated as the difference between the night after ketamine infusion and the baseline electroencephalogram sleep night. (b, left) The correlation between SWA and BDNF in responders (r = 0.595, p = 0.032) and non-responders (r = 0.215, p = 0.424) to ketamine. Note the change in y axis scaling between responders and non-responders. (b, right) The correlation between slow wave incidence and BDNF in responders (r = 0.626, p = 0.022) and non-responders (r = 0.033, p = 0.904) to ketamine.
Relationship between mood, BDNF, and slow wave sleep changes
In order to assess the clinical implication of the observed changes in both SWA and BDNF, we performed separate analyses for those patients whose depressive symptoms clinically improved after ketamine infusion (responders) vs. those whose symptoms did not improve (non-responders). No significant differences were observed between responders and non-responders with regard to baseline BDNF and SWA levels. The responder subgroup (n = 13) showed significant correlations between the EEG sleep variables [SWA (r = 0.595, p = 0.032) and slow wave amplitude (r = 0.626, p = 0.022)] and change in log BDNF. In contrast, these significant relationships were not observed in the non-responder subgroup (n = 16; Fig. 3b). Visual inspection of the top (n = 8) and bottom (n = 8) quartiles of the baseline BDNF score distribution indicated that the lower quartile had a greater incidence (χ2, p < 0.05) of responders (n = 6), all of whom exhibited large increases in BDNF levels after ketamine infusion. The upper quartile had more non-responders (n = 6) than responders, with more variability in BDNF change after infusion. Interestingly, the lower quartile showed greater increases in both BDNF levels and SWA compared to the upper quartile (t = 6.647, p < 0.001, df = 28; t = 2.975, p < 0.05, df = 28, respectively).
Discussion
This study is the first to describe the effects of ketamine infusion on EEG sleep slow waves and their relationship with plasma BDNF and improvement in depressive symptoms in patients with treatment-resistant MDD.
In particular, our results demonstrated that ketamine infusion rapidly (within hours) improved depressive symptoms in a group of drug-resistant patients with MDD and that several factors were associated with clinical improvement, including change in plasma BDNF levels (collected 230 min after ketamine infusion), change in SWA and amplitude/slope of individual slow waves (measured during the subsequent night of sleep). Furthermore, the changes in both SWA parameters and BDNF levels were found to be proportional specifically in responders – those patients whose depressive symptoms clinically improved after ketamine infusion. Consistent with previous studies from our laboratory (Machado-Viera et al.2009), no difference was noted in BDNF levels between responders and non-responders, although the relationship between surrogate markers of plasticity was strongest in the cohort of responders. The modest relative increase in BDNF at 230 min – a finding not seen in our previous study (Machado-Viera et al.2009) – may be related to the variability of BDNF levels in this treatment-resistant sample, the use of different analytical methods (e.g. log-transformed, change scores), as well as our theoretical focus on BDNF levels nearest to sleep onset. It is important to note that sleep slow wave biomarkers are functionally related to increased synaptic strengthening and cortical synchronization. Thus, the fact that the correlation between slow wave measures and BDNF change was limited to the cohort of ketamine responders suggests that ketamine-induced changes in BDNF result in proportional synaptic strengthening in ketamine responders.
It is interesting to note that increased hippocampal and cortical BDNF expression accompanied by antidepressant-like effects was found in recent studies investigating the effects of a single i.p. injection of ketamine in rats (Autry et al.2011; Reus et al.2011). The induction of neurotrophic factors may therefore partially underlie the antidepressant effects of ketamine, given that MDD is associated with low levels of BDNF and that chronic treatment with antidepressants elevates BDNF levels (Allaman et al.2011). BDNF is also a key neurotrophic factor involved in the orchestration of plastic changes leading to synaptic potentiation (Lu, 2003), including the recruitment of 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptors at the synapse. Converging lines of evidence suggest that ketamine's rapid antidepressant effects may occur via AMPA-mediated synaptic potentiation of neural circuits implicated in the pathophysiology of MDD (Maeng & Zarate, 2007), in line with a growing body of data indicating that synaptic plasticity is altered in mood disorders (Schloesser et al.2008). A similar potentiating effect on synapses has been observed in animal experiments following sleep deprivation, which increased AMPA receptor density in synaptoneurosomes from both cerebral cortex and hippocampus. Notably, this synaptic strengthening was reflected in an increase in the slope and amplitude of cortical evoked response and was subsequently correlated with increased sleep SWA (Vyazovskiy et al.2008). Another similarity between ketamine infusion and sleep deprivation is that both can bring about AMPA-mediated synaptic potentiation rapidly, whereas conventional antidepressants do so in a delayed manner through a cascade of intracellular signalling changes (Pittenger et al.2008).
Previous electrophysiological studies in rats also showed that peritoneal injections of ketamine (Feinberg & Campbell, 1993) induced changes in sleep SWA during NREM. Our results extend these observations in humans and suggest a link between a ketamine-induced increase in BDNF levels and enhanced SWA parameters in MDD patients, as indicated by the correlation between post-infusion sleep slow wave and plasma BDNF level changes.
The notion that ketamine-induced increases in BDNF may be accompanied by an increase in SWA and amplitude is further strengthened by several studies that directly examined the effect of BDNF on EEG sleep slow waves (Faraguna et al.2008; Huber et al.2007). Interestingly, these studies indicated that SWA was: (a) increased by intrahemispheric infusion of BDNF; (b) diminished by BDNF antagonism (Faraguna et al.2008); (c) increased by behavioural interventions that increase central levels of BDNF (Huber et al.2007) as well as the related plasticity-related genes Arc, Homer and NGFI-A (Huber et al.2007).
It is important to note that, prior to ketamine, sleep deprivation and electroconvulsive therapy (ECT) were the only therapeutic options for which a rapid (within a few days) antidepressant response had been convincingly demonstrated (Hemmeter et al.2010; Husain et al.2004; Pagnin et al.2004). Intriguingly, both sleep deprivation and ECT seem to increase the expression of neurotrophins, especially BDNF (Altar et al.2003; Vyazovskiy et al.2008) and result in an overall slowing of resting EEG activity (Hughes, 1996; Vyazovskiy et al.2008).
Although this study provides the first description of the effects of ketamine infusion on synaptic plasticity in patients with treatment-resistant MDD, several open questions and study limitations remain that will need to be addressed here and in future work. First, although consistent with the possibility that sleep slow wave changes observed in our study might be due to increased brain BDNF, our inability to directly measure brain BDNF, together with the fact that the association between blood BDNF and brain BDNF levels in humans is the subject of ongoing research (Sen et al.2008), suggests that careful interpretation of our findings is warranted. Preclinical studies report the passage of BDNF from blood to the central nervous system and vice versa (Pan et al.1998), consistent with the notion that peripheral and central measures of BDNF are highly correlated (Karege et al.2002b). Furthermore, while we cannot be certain of the peripheral vs. central origins of measured BDNF in this study, it is possible that ketamine also increases peripheral BDNF, followed by central active transport (for a discussion, see Sen et al.2008) and concomitant increased sleep slow wave (and mood) effects. This possibility would have important clinical and treatment implications. However, we are unaware of any studies that have examined the effects of peripherally administered BDNF on sleep slow waves.
Second, this study lacked a placebo control. While the assessment of antidepressant drug efficacy often requires a placebo comparison group, selecting an appropriate placebo to evaluate ketamine's effects on physiological markers such as sleep EEG and BDNF is particularly challenging. For example, should the placebo treatment be inert with regard to SWA or EEG spectral change or could an active placebo be a lower dose of the active treatment that targets similar molecular mechanisms but with subclinical effects? (Knott et al.2006, 2011). Selecting an appropriate active placebo that did not affect EEG spectra would be particularly difficult, given that many drugs alter EEG bands of interest (Gaillard & Tissot, 1975; Johnson et al.1976). Thus, it is likely that little scientific insight into ketamine's antidepressant mechanism of action would be gained by using a placebo control in the present study. Furthermore, whatever gain could be achieved would have to be weighed against the ethics of increasing the research burden on these drug-free, treatment-resistant MDD patients.
The primary objective of this investigation was to obtain neurophysiological and molecular evidence that ketamine increases synaptic efficacy. We capitalized on well-documented evidence of ketamine's antidepressant effects (see Bunney & Bunney, 2011) and concluded that one of the best controls for physiological measures associated with an antidepressant effect is an intra-individual comparison offered by the baseline condition, as used in our current analysis. Therefore, the rationale for our study design, which measures change in physiological parameters in clinically distinguishable patients/populations, offers an internal control for the efficacy of ketamine infusion. In addition to being an efficient way to conduct clinical research, the design thus had research strengths that directly took into account the large variation between patients.
Taken together, the results presented here suggest that sleep SWA parameters and BDNF serve as non-invasive indices for testing the efficacy of newly developed antidepressant therapies that target the glutamatergic system. The results also suggest that strengthening neuronal synapses may be a common mechanism underlying the rapid antidepressant effects of ketamine, thus providing novel insights in our quest to understand the neurobiology of MDD and to develop more effective diagnostic and therapeutic interventions.
Acknowledgements
Dr Duncan, Ms Selter, Dr Hejazi, Dr Yuan, Ms Brutsche, and Dr Zarate gratefully acknowledge the support of the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH), and thank the 7SE Research Unit of the NIMH-NIH for their support. Ioline Henter (NIMH) provided invaluable editorial assistance.
Statement of Interest
This study was supported in part by the IRP-NIMH-NIH. The author(s) declare that, except for income received from our primary employer, no financial support or compensation has been received from any individual or corporate entity over the past 3 yr for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. Drs Zarate and Manji are listed as co-inventors on a patent application for the use of ketamine in major depression. Drs Zarate and Manji have assigned their rights on the patent to the US Government but will share a percentage of any royalties that may be received by the government. Dr Tononi has consulted for Sanofi-Aventis and Takeda and is currently the David P. White Chair in Sleep Medicine at the University of Wisconsin Madison, endowed by Phillips Respironics. Dr Tononi has also received unrelated research support from Phillips Respironics. Dr Riedner is financially supported in part by the research funds given to Dr Tononi by Philips Respironics.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}