
Abstract
Background
Light chain (AL) amyloidosis is a rare, complex disease associated with significant morbidity and mortality. Delays in diagnosis are common and may have detrimental consequences on patients’ prognosis. Too little is known regarding the patient journey to diagnosis.
Objective
The objective of this study was to describe the patient-reported journey to a correct diagnosis for AL amyloidosis.
Methods
Using a mixed-methods approach, data were collected from clinician (n = 4) and patient (n = 10) interviews and a survey of community-based patients with AL amyloidosis (n = 341). Data were used to document the patient experience between the onset of symptoms and the receipt of a diagnosis.
Results
Delays in diagnosis were common. Qualitative and quantitative data indicated that initial symptoms were varied and similar to other more prevalent diseases. Two themes regarding the journey to diagnosis emerged: (1) barriers to an early diagnosis; and (2) the emotional toll of the journey. Time to diagnosis was heavily influenced by how patients interpreted their initial symptoms, whether they sought early medical help, and challenges associated with making differential diagnoses. Survey results indicate that patients with primary cardiac involvement were more likely to receive a delayed diagnosis than those with primary kidney involvement. Patients described mixed emotions associated with the eventual diagnosis of AL amyloidosis.
Conclusions
These data support a need for better early identification and support for patients seeking a diagnosis. Increasing clinician awareness may reduce the time to diagnosis. Additional research is needed to identify optimal diagnostic testing to reduce delays in treatment initiation and subsequent severe impacts on health.
Similar content being viewed by others

Patients with light chain (AL) amyloidosis often experience a complex, lengthy, and ambiguous journey to diagnosis, which can affect their emotional well-being. |
Older patients may delay seeking medical help because they attribute early, common symptoms, such as fatigue or changes in appetite, to the aging process or to other chronic, more common conditions. |
Clinicians face diagnostic challenges due to lack of awareness of AL amyloidosis, the non-specific symptoms of the disease, and the heterogeneity of the condition. |
1 Introduction
Light chain (AL) amyloidosis is a rare, heterogeneous condition that affects approximately 8–12 new individuals per million person-years [1, 2]. It is a complex and often fatal disease caused by misfolded protein deposits composed of monoclonal immunoglobulin light-chain fibrils in tissues and organs [3]. Amyloid deposits and circulating free light chains interfere with the structure and function of tissues and can lead to permanent organ damage and death [4, 5]. The primary risk factor associated with AL amyloidosis is increased age. The median age at diagnosis is 64 years [6].
Although the deposits can be localized to one specific tissue or organ, it is typically a multi-system disease. The locations of amyloid deposits can vary, thus contributing to the heterogeneity of the disease experience in terms of signs, symptoms, and health complications [7]. Heart and kidney involvement, the most commonly affected organs, can lead to renal failure, cardiomyopathy, and pericardial and pleural effusions [4]. Damage to multiple organs and cardiac impairment are most predictive of decreased survival rates and poor disease outcomes, with the most common cause of death being a cardiac complication [4].
Prognosis for AL amyloidosis is associated with early diagnosis, treatment, and the extent of organ involvement. Current treatments, including chemotherapy regimens, autologous stem cell transplants (ASCT), and immunomodulatory drugs, have been shown to improve prognosis and extend life expectancy [8]; however, no treatment has been specifically approved by USA or European regulatory agencies for the treatment of AL amyloidosis. Although current treatments aim to reduce the production of clonal plasma cells and to prevent further misfolded light chains and amyloid accumulation and deposition, there are currently no approved treatments to target or impact the existing amyloid that causes organ dysfunction. With current treatment strategies, successful treatment outcomes depend on early diagnosis and the extent of organ damage prior to treatment initiation [9]. In most cases, organ damage is irreversible; therefore, rapid treatment initiation is necessary to receive the optimal treatment benefit.
Despite previous reports of diagnostic delays and their detrimental impact on patient prognoses, there are limited data to describe patients’ journey to a diagnosis [10]. It is plausible that delays in diagnosis may be multi-faceted and heavily influenced by the average age of the patient, the complexity of the disease, and the rareness of the condition. The onset of disease often occurs at an age when individuals are at an increasing risk for other more common, chronic conditions. The complexity of the disease and the non-disease-specific symptoms may reduce the likelihood of a prompt diagnosis. Finally, first-line clinicians may not be familiar with such a rare condition and may not initially consider it when making decisions related to differential diagnoses.
An in-depth understanding of the diagnostic process from the perspective of both clinicians and patients may identify modifiable barriers to diagnosis and treatment initiation. The purpose of this study is to describe the journey to an AL amyloidosis diagnosis using data from qualitative and quantitative research and to raise awareness of the difficulties that patients encounter in order to obtain a correct and timely diagnosis of their disease.
2 Methods
This study was based on a mixed-methods triangulation approach—a method in which interpretations are made equally from qualitative and quantitative data sources [11]. Qualitative interviews were conducted with clinicians and patients to assess patients’ experience with the disease and to inform the development of a quantitative survey on the same topics. Both data collection efforts that involved patients were reviewed and approved by the New England Institutional Review Board (qualitative patient interviews: NEIRB #15-140; quantitative survey: NEIRB #15-355).
2.1 Qualitative Interviews
2.1.1 Recruitment and Procedures
Study investigators sought to recruit three to five clinicians from several key specialties and to yield an even distribution of clinicians by gender and representation from different geographic regions. Clinicians were eligible for the study if they had (1) at least 5 years’ experience in post-residency clinical practice and (2) expertise in treating patients with AL amyloidosis. Potential clinician respondents were identified and sent introductory emails by the study sponsor to inform them of the study opportunity. Semi-structured interviews with clinicians were conducted by telephone in March 2015. Data for this paper were drawn from portions of the interview that focused on (a) clinician experiences related to pathways to diagnosis and (b) initial signs and symptoms of the disease.
For patient interviews, investigators sought to recruit a sample of ten patients who varied in terms of gender, educational attainment, age, geography, duration of disease, types of organ involvement, and experiences with treatment success. Patients were eligible to participate if they (1) were at least 18 years of age; (2) had a self-reported diagnosis (as made by a doctor) of AL amyloidosis; and (3) were willing and able to participate in the study and comply with all study requirements, including completing a written informed consent form (ICF). Patients were excluded from the study if they were unable to answer questions by phone as required to participate in the study. The Amyloidosis Support Group, a patient advocacy group, sent an email to a random selection of 50 active group members. The email included an introduction to the study, a patient screener, the ICF, and instructions for returning the completed screener and ICF. The semi-structured interviews, conducted between April and May 2015, were designed to gather information related to patients’ journey to diagnosis, specific initial symptoms, and the impact of AL amyloidosis on their functioning or well-being.
2.1.2 Data Coding and Analysis
All interviews were audiotaped, transcribed, coded, and analyzed using NVivo Software (QSR International Pty Ltd., Melbourne, VIC, Australia, Version 10, 2012) and a grounded theory approach [12]; this approach allows themes to emerge from the data rather than imposing a priori hypotheses to be tested. Physician interviews were coded and analyzed by one researcher, whereas patient interviews were dual coded and analyzed by four researchers. Review meetings were held to ensure agreement between coders. An iterative process for coding was developed to demonstrate saturation, the point at which no new relevant information emerges and no additional interviews are necessary. First, a preliminary set of first-level codes was developed by reviewing all ten interviews. Next, the transcripts were divided into four groups. The first group of interviews was recoded for second-level codes. If new second-level codes emerged from the second batch of interviews, the first group of interviews was re-reviewed using the revised list of new codes. This process was continued with the third and fourth groups. Saturation was confirmed once the last interview was reviewed and no new codes emerged.
2.2 Quantitative Survey
A longitudinal, non-intervention study of community-based patients with AL amyloidosis was conducted to examine the experience of patients with AL amyloidosis and the impact of the disease on their health-related quality of life (ClinicalTrials.gov identifier NCT02574676). Investigators partnered with two patient advocacy groups (the Amyloidosis Support Groups and the Amyloidosis Foundation) to post recruitment announcements on websites and social media pages, as well as in membership emails. Individuals were deemed eligible for the study if they (1) were at least 18 years of age; (2) reported a diagnosis of AL amyloidosis made by a physician; and (3) were able and willing to complete four online surveys written in English during a 12-month follow-up period. Data for the cross-sectional analyses reported here were drawn from the initial survey using a subset of survey items pertaining to basic demographics, disease characteristics, initial symptoms, and the patients’ journey to a diagnosis.
Descriptive statistics were calculated using SAS® version 9.2 (SAS Institute Inc., Cary, NC, USA). Chi square tests were used to conduct bivariate analyses. Logistic binomial models were used to estimate the relative risk of receiving a delayed diagnosis, defined as a diagnosis that occurred at least 6 months after the onset of initial symptoms.
3 Results
3.1 Study Participants
Demographic characteristics of the clinician (n = 4: three hematologists and one nephrologist) and the demographic and disease characteristics of the patient interviewees (n = 10) and survey respondents (n = 341) are described in Table 1. Both patient samples were diverse in terms of geographic representation. For example, patient interviewees and survey respondents came from all regions of the USA with respondents residing in 40 states.
3.2 Journey to Diagnosis
Delays in obtaining a diagnosis of AL amyloidosis were commonly reported by both clinicians and patients. According to clinicians, the timeframe between symptom onset and the receipt of a diagnosis was 10 months (range 1 month to 2 years). Patient interviewees also reported delays in time to diagnosis that varied broadly (average 3 years; range 3 months to 4 years). This was further corroborated by the survey data where over 70% of patients reported that they received a diagnosis at least 6 months following the onset of symptoms (Table 2).
There was wide variation in how and when patients received their AL amyloidosis diagnosis. For some, the path to a diagnosis was straightforward; whereas, for others, the experience involved misdiagnoses and many consultations with various specialists. For example, one patient interviewee was diagnosed within 3 months of receiving an abnormal urine test during a routine physical examination. In contrast, another patient’s journey took more than 4 years from the time he/she first sought help from a doctor for a bothersome symptom to when he/she ultimately received a confirmed diagnosis.
3.2.1 Barriers to an Early Diagnosis
Patients and clinicians reported cumulative factors that ultimately led to delays in diagnosing AL amyloidosis. Patients’ interpretation of the initial signs and symptoms of the disease influenced whether they sought early medical help. Once patients began to seek a diagnosis, delays also occurred within the healthcare system. Clinicians were not often familiar with the condition and were challenged by the complexity and rarity of the disease. This impacted their ability to make a differential diagnosis. It is important to note that given the variability of the disease, there was no “typical” case of AL amyloidosis, and the variability of the disease itself contributed to delays in reaching a diagnosis.
3.2.1.1 Interpretation of Signs and Symptoms by Patients Seeking Medical Help
Based on spontaneous reports and targeted probing questions, patient interviewees reported a total of 32 distinct symptoms, regardless of type of organ involvement. The symptoms most commonly reported during the interviews were used to generate close-ended questions on the quantitative survey. As shown in Fig. 1, the most prevalent symptoms were consistent across both data sources. Fatigue, the most commonly reported symptom, was reported by 80% of all patient interviewees and 82% of all survey respondents. Overall, many of the reported symptoms were non-specific and overlapped with the symptomology of other more prevalent chronic conditions, such as cardiovascular disease or cancer.

Prevalence of commonly reported initial symptoms among patients with light chain amyloidosis
These non-specific, early symptoms did not always raise immediate concern for patients, and may have contributed to delays in seeking medical attention. It was common for older patients to associate initial symptoms with the aging process or to attribute them to other existing health conditions. For some patients, the early symptoms were minor and only seemed relevant after they had received the diagnosis. Three of the ten patient interviewees did not consider they were seriously ill until they received an abnormal result on a routine urinalysis despite noting previous symptoms as illustrated below:
“I’ve always had problems with statins, so I’ve always had joint and muscle pain, because statins have always bothered me. So, I didn’t think much about it when my hands started tingling and I had neck pain.” (Patient 003)
“It was things like being a little more tired, and I was suddenly, and I don’t know how this fits in, but I had been a vegetarian or semi-vegetarian for quite some time, and all of a sudden I would have these horrible cravings for meat, and I thought it had something to do with iron, being of a certain age or whatever.” (Patient 010)
“As part of that whole screening process [screening for eligibility for a stem cell transplant] questions were asked did you have any experience with this or that or the other thing. Then it became evident to me I was seeing some minor symptoms. Nothing I would have said caused me to go to see a doctor.” (Patient 002)
For some patients, the initial symptoms were more severe and the negative impact on daily functioning was readily apparent. This may be particularly true for younger patients whose initial symptoms may not have fit the expectancies of their current health status.
“And it was unusual. I never had shortness of breath. And then it wasn’t every single time. You know, I’d also notice things like I’d finish dinner and I’d go out and play catch with my son and I’d get out of breath. And it was a bunch of unusual circumstances like that. And finally I – the final thing that got me into the doctor was I went for a walk with my daughter, made it about a half mile and had to stop and rest. Like, okay, that’s definitely not normal.” (Patient 004)
3.2.1.2 Patient Characteristics Associated with Delayed Diagnoses
Table 3 describes the patient characteristics of the survey respondents by their time to diagnosis. In this sample, patients who received an early diagnosis did not differ in terms of basic demographics from patients who received a delayed diagnosis. Among patients who received delayed diagnoses, greater proportions reported non-disease-specific symptoms, such as fatigue, shortness of breath, a feeling of fullness in the stomach, dizziness upon standing, and numbness of arms and legs. Symptoms such as purpura, which are less common in other prevalent diseases, were not associated with time to diagnosis.
There were also significant associations between the primary organ affected and the time to diagnosis based on bivariate analyses. For instance, 35% of patients who received a delayed diagnosis identified heart as the primary organ affected by the disease; whereas only 19% of patients who received an early diagnosis identified their heart as their most affected organ. Patients who identified the heart as their most affected organ were 43% more likely to obtain a delayed diagnosis as compared to patients who identified the kidney as their most affected organ (relative risk [RR] 1.43, 95% confidence interval [CI] 1.21–1.69). The risk of a delayed diagnosis among patients who identified other types of primary organ involvement (i.e., liver, gastrointestinal, and nervous system) was similarly 42% greater than the risk among the patients with primary kidney involvement (RR 1.42, 95% CI 1.20–1.68).
3.2.1.3 Challenges of a Differential Diagnosis
Once patients began to actively pursue a diagnosis due to either (1) suspicious or bothersome symptoms and/or (2) abnormal findings from a routine test, the path to a diagnosis was heavily influenced by the interpretations of the signs and symptoms by clinicians. Clinicians described the journey to a differential diagnosis as challenging, noting that if patients present with symptoms that were more specific to AL amyloidosis, such as macroglossia (i.e., enlarged tongue) or pinched periorbital purpura, that could facilitate the diagnostic process; however, few patients have these symptoms. The vast majority of patients present with non-specific symptoms such as fatigue. Clinicians acknowledged that this presents a challenge for first-line clinicians. One clinician (female, hematologist) used the term “a great masquerader” to describe AL amyloidosis because its symptoms are also common to other, more prevalent conditions.
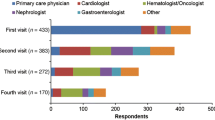
Patients in both the qualitative and quantitative study visited multiple doctors during their journey to a diagnosis. Among surveyed patients, more than a quarter (28%) saw at least six doctors across various specialties before they ultimately received their AL amyloidosis diagnosis (Table 2). Patient interviewees consulted with an average of three different types of specialists before they received the correct diagnosis. As described in Table 4, the types of specialty consultations depended on the patients’ organ involvement and included primary care physicians, cardiologists, hematologists, nephrologists, gastroenterologists, pulmonologists, and others.
Patients’ diagnostic narratives described many examinations and tests. Even after AL amyloidosis was suspected, additional tests were conducted for confirmation and to further determine which organs were affected. During interviews, patients spontaneously described a broad range of diagnostic tests that included urinalysis and blood tests, biopsies, and various imaging procedures. Although patient interviewees were not systematically queried about specific types of diagnostic tests, all interviewees reported receiving at least one biopsy (average number of biopsies = 2; range 1–4). Bone marrow biopsies were the most frequently mentioned (70% of interviewees). Other biopsy locations included fat pad, heart, lung, kidney, skin, and upper/lower gastrointestinal tract. Aside from biopsies, patients also received the following diagnostic tests or procedures: bronchoscopies; cardiac catheterizations; cardiac magnetic resonance imaging; colonoscopies; computerized tomography scans; echocardiograms; electrocardiograms; endoscopies; nerve conduction tests; positron emission tomography scans; pulmonary functioning tests; and X-rays.
Clinicians further confirmed that delays in treatment initiation occur due to the diagnostic process and additional diagnostic testing often occurs even after a patient is referred to a specialty clinic or a center of excellence. Based on these interviews, clinicians reported that it can take 1–2 months to receive confirmation of their diagnosis.
Misdiagnoses may also prolong patients’ diagnostic journey. Eight of the ten patient interviewees received incorrect diagnoses prior to receiving a conclusive AL amyloidosis diagnosis.
“He started talking about; he didn’t say amyloidosis at that time but he said you know could be some variant of leukemia in your blood system. Could be myeloma.” (Patient 002)
“At that point the diagnosis was still cardiomyopathy … hypertrophic cardiomyopathy. When I was in the hospital to get the defibrillator pacemaker a team came in and basically just shook their heads and said, you know, something’s not right with this diagnosis so we’re going to do a biopsy. They did a biopsy and the next day installed the defibrillator. I went back for a check on the silver bandage the week after that where they advised me that I had cardiac amyloidosis.” (Patient 005)
“And when she came back, she said, ‘Oh, you have got something called pre-leukemia, MDS, myelodysplastic syndrome.’ She said that ‘That’s MDS, which is a form of cancer.’” (Patient 008)
“I think the asthma [diagnosis] absolutely was incorrect and going through all of the asthma medications, of course, did not help.” (Patient 010)
3.2.2 Emotional Toll of the Diagnostic Process
Reactions to receiving a diagnosis of AL amyloidosis were mixed. Many patients expressed a feeling of relief after receiving their ultimate diagnosis, due to the anxiety and uncertainty of symptoms that could not be explained. Other patients were shocked and overwhelmed by the rareness and seriousness of the condition. The following quotes are representative of reactions described by patients:
“… the longer it went the more and more frightened I got because they couldn’t identify it … when they finally said it’s amyloidosis, it’s almost a relief because I finally knew what it was.” (Patient 001)
“At the same exact moment [when I received the diagnosis] I feel relieved that actually I had a diagnosis, that I wasn’t crazy. I mean there was something wrong with me that wasn’t medically identifiable because, if not relief, there was a certain sense of justification that it is all good. And then it hit me when they started saying, yes, but there is no cure for AL.” (Patient 008)
“I can tell you the exact date when it happened, I know it was probably about 4:00 and I remember exactly where I was standing because I was at work, and I had gone into a stairwell to take his call, and I can remember in detail most of the conversation so in terms of feelings, it’s not exactly a feeling but it was life-changing, I knew that. I remember being distressed and wondering really what was going to happen to me, and it was a little bit of a shock, so I didn’t necessarily cry or anything at that moment, I just remember being kind of overwhelmed, which is probably the word.” (Patient 010)
4 Discussion
This rigorous mixed-methods study documents important aspects of the journey to diagnosis from both the clinician and patient perspective. Based on qualitative and quantitative data, delays in obtaining the correct diagnoses for AL amyloidosis occur often. Common themes observed in patient journeys included the barriers to an early diagnosis and the emotional toll associated with both the disease and the process.
Patients were not always concerned by initial symptoms. Since AL amyloidosis is more likely to occur later in life, it is not surprising that some of the non-specific symptoms, such as fatigue and loss of appetite, were misinterpreted as symptoms of aging or attributed to more common chronic ailments. Interestingly, purpura, a symptom that may be more distinctive of this disease, was not significantly associated with time to diagnosis in bivariate analyses. Though purpura is a distinctive symptom of the disease, its prevalence in patients with AL amyloidosis is estimated to be less than 15% [13]. Thus, first-line clinicians, who may have little experience with AL amyloidosis, may still be relatively unfamiliar with the diagnostic utility of this symptom. Indeed, recent work has described cases in which a proper diagnosis is missed even in the presence of this symptom [14]. In working with clinicians to reduce barriers to a prompt and correct diagnosis, the importance of this symptom should be emphasized. It is also important to note that these symptoms are varied and do not occur in isolation, making it difficult to tease out whether specific symptoms may serve as early indications of the disease and lead to earlier diagnoses.
Several patients were first alerted by abnormal results on routine urine tests rather than by their specific symptoms. Results from the quantitative study further indicated that patients with kidney involvement were less likely to experience the diagnostic delays incurred by patients primarily affected by other types of organ involvement. It is plausible that when patients receive abnormal urinalysis results they may be more likely to see a nephrologist who will order a kidney biopsy much earlier in the diagnostic journey than other patients. This may subsequently accelerate diagnoses for patients with kidney involvement even if a specialist does not suspect or is even aware of AL amyloidosis. Further examination of whether patients with particular symptom profiles, disease characteristics, or patterns of healthcare utilization and proximity to a center of excellence (i.e., individuals who see their doctors regularly and/or adhere to recommended health screenings) are more or less likely to receive a delayed diagnosis is warranted. Additional studies aimed at understanding how clinicians make diagnostic decisions may have the potential to increase the accuracy and speed with which first-line clinicians are able to recognize the disease.
Patients described a variety of tests and procedures used to confirm their diagnosis and/or to identify affected organs. Additional research should be conducted to identify optimal diagnostic strategies based on sensitivity, specificity, and low risks for complications. Unnecessary biopsies and the use of diagnostic testing with low sensitivity can lead to delays in diagnosis and treatment initiation. A recent study reported a median delay in diagnosis of 4.1 months in cases that required more than one biopsy as compared to patients who only needed one biopsy to obtain a confirmation of their diagnosis with AL amyloidosis [10].
Nearly three-quarters (72%) of patients in the quantitative study received a diagnosis at least 12 months after the onset of symptoms. This high prevalence of delayed diagnoses suggests that the overall prevalence of the disease may be underestimated. Many patients may die of a complication before receiving a diagnosis. Delays in diagnosis for cardiac patients are especially concerning, as the median survival rate among untreated patients with cardiac involvement is approximately 6 months following the onset of symptoms [6]. Furthermore, the prognosis for patients with AL amyloidosis is improved by early diagnosis and treatment—the challenges and delays described by both clinicians and patients have the potential for a severe impact on health outcomes and impaired quality of life. These data support a need for better early identification and support for patients looking for a diagnosis. Clinical experts recommend the inclusion of AL amyloidosis as a potential diagnosis when patients present with nephrotic-range proteinuria, unexplained non-ischemic cardiomyopathy, peripheral neuropathy, unexplained hepatomegaly, or atypical multiple myeloma [8]. Heightened awareness and understanding of the disease and identification of the optimal coordination of care is paramount to help patients receive a correct diagnosis, receive prompt treatment, and manage their disease effectively. Unnecessary procedures and misdiagnoses not only delay treatment, but may negatively impact the patient-physician relationship and increase healthcare utilization and subsequent costs. Data collected from the longitudinal study may be useful in more clearly illustrating the relationship between time to diagnosis and future health outcomes.
A number of study limitations should be noted. First, interviews with clinicians were conducted with experts who treat AL amyloidosis regularly. Their reports may not fully capture some of the challenges or experiences of first-line clinicians who see patients earlier in the diagnostic journey and are less familiar with the disease. Similarly, patients who are able to participate in a long, in-depth interview or complete an online survey may not be representative of the broader disease population. Given the expectations to participate in the study, patients who are too ill may not have agreed to participate. This, and other potential selection biases, may have impacted observed patterns in the data, particularly if healthier patients tend to have similar diagnostic characteristics or histories. Despite these limitations, these findings consistently highlighted common barriers to diagnosis and patient experiences.
5 Conclusion
Patients experience a variety of challenges during their journey towards a diagnosis of AL amyloidosis. Increased awareness of the disease and its symptoms and clinical manifestations may reduce diagnostic delays, which could increase access to treatment and ultimately improve health outcomes.
References
Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817–22.
Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SD, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161(4):525–32.
Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337(13):898–909.
Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29(14):1924–33.
Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107(10):3854–8.
Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45–59.
Gertz MA. Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am J Hematol. 2013;88(5):416–25.
Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1:1331–41.
Palladini G, Milani P, Foli A, Obici L, Lavatelli F, Nuvolone M, et al. Oral melphalan and dexamethasone grants extended survival with minimal toxicity in AL amyloidosis: long-term results of a risk-adapted approach. Haematologica. 2014;99(4):743–50.
Sayago I, Krsnik I, Gómez-Bueno M, García-Pavía P, Jaramillo N, Salas C, et al. Analysis of diagnostic and therapeutic strategies in advanced cardiac light-chain amyloidosis. J Heart Lung Transplant. 2016;35(8):995–1002.
Creswell JW, Plano Clark VL. Designing and conducting mixed methods research. 2nd ed. Thousand Oaks: Sage; 2011.
Strauss AL, Corbin JM. Basics of qualitative research: grounded theory procedures and techniques. Newbury Park: Sage Publications; 1990.
Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121(26):5124–30.
Colucci G, Alberio L, Demarmels Biasiutti F, Lämmle B. Bilateral periorbital ecchymoses. An often missed sign of amyloid purpura. Hamostaseologie. 2014;34(3):249–52.
Acknowledgements
The authors thank the patients for their participation in this study and the Amyloidosis Support Group and the Amyloidosis Foundation for their help in patient outreach. Copy editing and administrative assistance were provided by Alexia Ng. OptumInsight Life Sciences, Inc. received financial support from Prothena Biosciences Inc. to conduct all data collection, analyses, and manuscript preparation for this study.
Author information
Authors and Affiliations
Contributions
MKW and MSB designed the qualitative study and collected/coded the qualitative data. IL and MF contributed to the discussion guides and recruitment of patients. KLM, MKW, MSB, and SDG designed the quantitative study and collected data. KLM analyzed the data. KLM, MKW, MSB, SDG, and TQ contributed to the conceptualization and writing of the manuscript drafts. All authors revised the manuscript critically for important intellectual content and gave final approval of the version to be published.
Corresponding author
Ethics declarations
Conflict of interest
Data collection and the writing of this manuscript were funded by Prothena Biosciences. S. Guthrie and T. Quock are employees of the study sponsor, Prothena Biosciences Inc. K. McCausland, M. White, and M. Bayliss are employees of OptumInsight Life Sciences, Inc. and received research funding from Prothena Biosciences Inc. to conduct the study. M. Finkel and I. Lousada do not have any conflicts of interest to declare.
Ethical approval/informed consent
Both studies were approved by the New England Institutional Review Board (Qualitative patient interviews: IRB #15-140; Quantitative survey: IRB #15-355). All patients who participated in the qualitative interviews or the online survey provided written or digital informed consent, respectively.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McCausland, K.L., White, M.K., Guthrie, S.D. et al. Light Chain (AL) Amyloidosis: The Journey to Diagnosis. Patient 11, 207–216 (2018). https://doi.org/10.1007/s40271-017-0273-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40271-017-0273-5