
Abstract
Pseudobulbar affect (PBA) is a dysfunction of emotional expression characterized by involuntary outbursts of crying or laughing disproportionate or unrelated to mood, occurring in patients with various underlying neurologic disorders. This review describes the clinical data supporting dextromethorphan (DM) hydrobromide combined with quinidine sulfate (Q) as treatment of PBA and briefly surveys the ongoing debates concerning the terminology for dysfunction of emotional expression, as well as the ongoing searches for its brain substrates. Until recently, pharmacologic intervention consisted chiefly of off-label antidepressants. In October 2010, however, DM/Q at 20/10 mg twice daily received approval from the United States Food and Drug Administration for PBA in any setting, and in June 2013, dosages of 20/10 and 30/10 mg twice daily (labeled as 15/9 and 23/9 mg, respectively, DM/Q base) received approval from the European Medicines Agency. DM is an uncompetitive N-methyl-d-aspartate (NMDA) glutamate receptor antagonist, a sigma-1 receptor agonist, and a serotonin and norepinephrine reuptake inhibitor. To block DM hepatic metabolism, thereby increasing DM bioavailability, Quinidine, a cytochrome P450 2D6 inhibitor, is coadministered at a dosage well below those for treating cardiac arrhythmia. Three large-scale DM/Q trials have utilized PBA-episode counts and the Center for Neurologic Study-Lability Scale (CNS-LS), a validated PBA rating scale, to measure efficacy. In a 4-week study of patients with PBA in amyotrophic lateral sclerosis (ALS), DM/Q 30/30 mg was superior to its component drugs. A 12-week, double-blind, placebo-controlled study of DM/Q 30/30 mg showed similar efficacy in patients with PBA in multiple sclerosis (MS). A subsequent 12-week study of patients with PBA and ALS or MS showed superiority to placebo for the 20/10 and 30/10 mg doses. Efficacy was maintained during a 12-week, open-label extension (30/10 mg dose), with further improvement of mean CNS-LS scores. Across these studies, DM/Q was generally safe and well tolerated, with no evidence of clinically relevant cardiac or respiratory effects. DM/Q is being studied (currently unapproved) for conditions including agitation in autism and in dementia.
Similar content being viewed by others

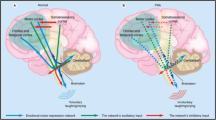

Introduction
Pseudobulbar affect (PBA) is a neurologic disorder of emotional expression characterized by frequent, sudden, involuntary outbursts of crying and/or laughing disproportionate or unrelated to the patient’s underlying mood, occurring in settings of neurologic disease or injury [1, 2]. Commonly associated conditions include amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), traumatic brain injury, stroke, Alzheimer’s disease, and Parkinson’s disease (PD) [1, 2]. Across these primary diagnoses, PBA prevalence estimates have been highly divergent [3], including values ranging from 5% in PD [4] to as much as 49% in ALS [5]. In a large-scale United States (U.S.) survey utilizing the two leading diagnostic tools, the Pathological Laughter and Crying Scale (PLACS), as validated in post-stroke patients [6], and the Center for Neurologic Study-Lability Scale (CNS-LS), validated in ALS [7] and in MS [8], the overall PBA symptom prevalence among patients with various primary diagnoses was 9.4–37.5%, depending on choice of tool and threshold value, implying a U.S. total of 1.8 to 7.1 million cases [3]. Owing in part to the potential for socially disruptive episodes, PBA may be embarrassing and distressful [9]. In a follow-on to the aforementioned survey, patients with PBA symptoms had significantly lower scores for general health, quality of life and relationships, and work productivity, compared with patients who had the same primary illness (weighted for severity) but not PBA [10]. PBA symptoms were also found to be an important reason for becoming housebound or suffering other adverse life situations. An accompanying video (see Video 1 in the electronic supplementary material) shows the intensity of a severe episode and, by implication, its psychosocial impact.
Until recently, pharmacologic intervention in PBA consisted chiefly of off-label use of antidepressants, on the basis of generally beneficial effects demonstrated in randomized, placebo-controlled, double-blind clinical trials of the tricyclic agents imipramine [11] and nortriptyline [6] in patients with underlying stroke; the tricyclic agent amitriptyline in patients with MS [12]; and the selective serotonin reuptake inhibitors (SSRIs) citalopram [13], fluoxetine [14, 15], and sertraline [16, 17], all in patients with stroke. Table 1 summarizes the methodology of these studies, selected for having been published in English-language medical journals after 1979. Overall, reported responder rates have often been higher for SSRIs than for tricyclics [18], and benefit has often been seen more rapidly and at a lower dosage than in the drugs’ on-label use for mood disorders [19], suggesting differing pathophysiologies or modes of action. Nevertheless, the findings derive mostly from trials hampered by small sizes, suboptimal designs, and limitations in defining suitable subjects and assessing improvement [18]. Among the aforementioned trials, only two [15, 17] had more than 28 subjects and only one [6], with 28 subjects, used a validated scale (i.e., PLACS). In a Cochrane Collaboration analysis [20] of five of the trials [6, 14–17], outcomes were judged to be consistent with large treatment effects, but with confidence intervals wide enough in three of the trials [6, 14, 16] to include the possibility of small positive effects or even small negative ones [16]. Off-label PBA interventions with less supportive evidence than has been published for antidepressants include the PD therapies levodopa and amantadine [21], the antiepileptic agent lamotrigine [22], the novel glutamate receptor antagonist ifenprodil [23], and thyrotropin-releasing hormone [24].
In October 2010, dextromethorphan (DM) hydrobromide administered in combination with quinidine sulfate (Q) at a dosage of 20/10 mg twice daily [25] received approval from the U.S. Food and Drug Administration (FDA) for PBA in any setting, on the basis of large-scale randomized, controlled studies of PBA secondary to ALS [26, 27] or MS [27, 28] (see Table 1). In June 2013, dosages of 20/10 and 30/10 mg twice daily (labeled by weight of the DM/Q base, at 15/9 and 23/9 mg, respectively) received approval from the European Medicines Agency (EMA) for PBA in any setting. In the pivotal DM/Q trial, the two regimens had been similarly effective for reducing PBA-episode rates [27]. DM is an uncompetitive antagonist of the class of glutamate receptors defined by their capacity to bind NMDA [29]. The drug is also known to be a sigma-1 receptor agonist [30] and serotonin and norepinephrine reuptake inhibitor [31]. Because sigma-1 receptor ligands including DM appear to protect neurons from glutamatergic excitotoxicity at least in vitro [29, 32], DM may have multiple antiglutamate properties. To block first-pass hepatic DM metabolism [33], DM is combined with Q, a potent cytochrome P450 2D6 enzyme (CYP2D6) inhibitor [34]. At 10 mg, Q achieves the required inhibition [35] at a dosage well below that required for a type 1a cardiac antiarrhythmic effect and, therefore, has only minimal potential for prolonging the QT interval [36, 37].
This review describes the clinical data supporting DM/Q as treatment of PBA. For context, it briefly surveys the ongoing debates concerning the terminology for dysfunction of emotional expression, and also the ongoing searches for its brain substrates. This review article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by the author.
Nosological Problems
The possibility of a relationship between neurologic insult and disordered emotional expression has been appreciated since at least the mid-nineteenth century, when Charles Darwin noted that “certain brain diseases, such as hemiplegia, brain-wasting, and senile decay, have a special tendency to induce weeping” [38]. Hermann Oppenheim described cases in which the disorder occurred amid recognized features of pseudobulbar palsy [39], evidently as a further capacity of bilateral forebrain lesions to mimic brainstem (bulbar) dysfunction. In 1924, S.A.K. Wilson proposed that the specific dysfunction was a motor disinhibition resulting when bilateral corticobulbar lesions uncouple a brainstem “faciorespiratory center” from cortical levels of control [40].
In ensuing years, Oppenheim’s term, “pseudobulbar affect,” and Wilson’s term, “pathological laughing and crying” (PLC), were joined by numerous others, including emotional lability, emotionalism, emotional incontinence, and pathological affect [41, 42]—a plethora that may inadvertently blur the distinction between disorders of emotion as a mental experience (i.e., mood disorders) and those disrupting the triggering of emotional expression as a motor program [43]. Convinced that the PLC he observed in ALS patients with progressive bulbar palsy was “a purely motor phenomenon involving the muscles of expression” [44], Klaus Poeck proposed criteria distinguishing PLC from emotional lability. In bulbar palsy, the episodes were stereotyped, uninterruptable, spontaneous (or triggered by nonspecific stimuli), and independent of mood, whereas in emotional lability, the episodes would be variable, interruptible (e.g., by distracting the patient), generally appropriate, but disproportionate, to their stimuli, and congruent to mood [45].
Most recently, in 2006, the term “involuntary emotional expression disorder” (IEED) has been advocated as an umbrella designation for conditions with “involuntary outbursts of crying and/or laughing” as their core feature [42]. IEED is seen as encompassing PLC and emotional lability, with PLC involving the disinhibition of motor programs for crying or laughing while emotional lability may be a more complex dysfunction of both mood and its expression [46]. For its part, the term PBA has been used both restrictively, to designate cases in patients with pseudobulbar palsy, and more broadly, like IEED, to encompass PLC and emotional lability [2]. Throughout what follows, descriptions of published research will retain the researchers’ choice of terminology. Elsewhere in this review, the term will have its broad meaning (reflecting the FDA and EMA approvals of DM/Q for treatment of PBA with no restriction of its neurologic setting).
Undoubtedly, the availability of competing, perhaps overlapping terms has hampered efforts to categorize disorders of emotional expression, ascertain their prevalence, and assess the evidence for therapeutic interventions [18]. But this is not to say that such difficulties arise solely from the terminology, and not the disorders themselves. In a study of 30 post-stroke patients with “emotionalism,” defined as having cried once or more during the preceding month, only five reported spontaneous episodes, only five reported no change in mood during episodes (although another 8 could not describe their mood), and only five felt they had no control over their crying [47]. Likewise, in a study of 21 ALS patients with PLC defined by a CNS-LS score ≥13, only two failed to have episodes triggered by stimuli “that might induce crying or laughing in anyone” [48]. In these patients, however, the episodes were “high-intensity, uncontrollable outbursts.” Moreover, the patients exhibited an impaired capacity to “hide your feelings” during emotion-eliciting film clips, further supporting theories that PLC is a dysregulation of emotional expression, as contrasted with a generalized hyperactivity in brain emotional systems.
Neuroanatomy and Pathophysiology
Efforts to uncover the brain substrates of PBA have relied on both neuroanatomic and neurophysiologic research techniques. The neuroanatomic research—in essence, the effort to correlate PBA with specific brain lesions, as identified at autopsy or most recently by neuroimaging—has been impeded not only by nosological problems but also the capacities of some of the primary insults underlying PBA to cause widespread damage. In a recent review of the neuroanatomical literature, the available evidence was judged to implicate prefrontal and anterior cingulate cortices and the internal capsule, thalamus, subthalamic nucleus, basis pontis, and cerebellum [49]. Among these loci, the basis pontis, a relay center for cerebellar afferents, was described as the only known site at which a discrete lesion can be sufficient to cause PLC.
Neurophysiologically, the classic source of evidence has been electrical stimulation. In humans, case reports have linked stimulation of the subthalamic nucleus to episodes of crying [50] and stimulation of cingulate cortex to episodes of laughter [51], each without change in mood. Episodes of crying or laughter are also well recognized to be prodromal features of seizures in hypothalamic hamartomas (dacrystic and gelastic epilepsy, respectively) [49]. In a set of five such patients, three had additional cortical seizures involving the cingulate gyrus [52]. The most recent neurophysiologic approach, applied directly to PBA patients, has been the recording of event-related potentials. In one set of experiments [53], 11 MS patients with PBA and 11 healthy controls heard a series of names selected for being subjectively significant or neutral to each of the subjects. Scalp electrodes recorded transient brain-tissue voltage waveforms. In general, the evoked current densities were greater in the MS/PBA patients than in the controls, both in sensory cortex and at later stages of cortical processing. For the neutral as well as the significant names, activation of motor areas was significantly greater in the MS/PBA group, implying disinhibition of a “gate-control” mechanism controlling emotional expression.
The available clinical evidence has prompted several detailed neuroanatomical hypotheses. In one proposal [54], PLC represents lesions in cortico-ponto-cerebellar pathways, impairing cerebellar capacities to adjust “the execution of laughter or crying” to the context of potential stimuli. In a more recent proposal encompassing IEEDs [46], a volitional pathway involving frontoparietal corticopontine projections is normally capable of inhibiting an emotion pathway involving frontotemporal projections to a complex comprising the amygdala, hypothalamus, and parts of the dorsal brainstem, which in turn coordinates the motor patterns of emotional display. In this view, lesions of the volitional pathway can result in PLC, while direct activation of the emotional pathway can result in emotional lability (or in the crying or laughter of dacrystic or gelastic epilepsy).
In principle, any of the numerous neurotransmitters thought to participate in mediating emotional expression might be important in PBA [2], including serotonin, glutamate, norepinephrine, dopamine, acetylcholine, gamma-aminobutyric acid, adenosine, corticotrophin-releasing hormone, and corticosteroids [43, 46]. Among them, serotonin is notable because of the benefits of SSRIs across a range of psychiatric disorders [55], and glutamate for its status as the major central nervous system (CNS) excitatory neurotransmitter [56].
DM/Q Efficacy
The in vitro capacity of DM to protect neurons from glutamatergic excitotoxicity [29, 32] was the initial impetus for clinical trials of DM’s potential benefit in neurodegenerative disorders, including unsuccessful efforts to identify neurophysiological improvement [57] or disease modification in ALS [58]. However, in studying DM as monotherapy (at up to 300 mg/day [57] or 1.5 mg/kg/day [58]), the trials neglected the drug’s hepatic metabolism by CYP2D6 [33]. In research exploring the extent of this conversion, plasma DM concentrations obtained in ALS patients 12 h after the final dose of a week of oral treatment at 120 mg/day were found to be “extremely low” (5 to 40 ng/mL) [34].
In clinical studies specifically of PBA, DM has been coadministered with Q (for CYP2D6 inhibition to increase DM bioavailability). The intent was to confirm early observations of improved emotional control in some ALS patients who received DM alone, and investigate the hypothesis of greater benefit if adequate CNS concentrations of DM are achieved by blocking its metabolism [59]. In a pilot crossover trial [60], 12 patients with “affective lability” in ALS received a month of DM/Q 30/70 mg and a month of placebo in a double-blind, randomized sequence, with each treatment taken once daily for 5 days and twice daily thereafter. Efficacy was assessed primarily by change in total score on a 65-item self-report emotional-lability questionnaire [59], which was later condensed and validated as the 7-item CNS-LS [7, 8]. By this measure, DM/Q showed significant benefit versus placebo. Intriguingly, DM/Q was also associated with significant reductions in episodes of anger and frustration, as assessed by an 8-item questionnaire subscale [61]. Such findings have become an impetus for further investigation (see below).
Three large-scale clinical studies of DM/Q for PBA [26–28] have all used the CNS-LS. Of the tool’s seven items, three address crying (e.g., “I find myself crying very easily”), and four address laughter (e.g., “I find that even when I try to control my laughter I am often unable to do so”). Each item is scored from 1 for “never” to 5 for “most of the time,” yielding a total score of 7 (for asymptomatic) to 35 (for worst symptoms). In its validation studies, use of a cutoff score ≥13 yielded accurate predictions of clinically detectable PBA in 82% of 77 ALS patients [7], and a cutoff of ≥17 yielded accurate predictions in 89% of 90 MS patients [8].
For a 28-day randomized, double-blind study of PBA in ALS [26], all subjects had a clinical diagnosis of PBA and a baseline CNS-LS score ≥13. Subjects were also required to have a baseline Hamilton Depression Rating Scale [62] score ≤16 (the threshold for moderate depression), and were excluded for any history of major psychiatric disturbance or current antidepressant medication use. During the study, 70 subjects received DM/Q 30/30 mg twice daily, 33 received DM 30 mg twice daily as monotherapy, and 37 received Q 30 mg twice daily. Per study protocol, the 11 subjects with a poor DM-metabolizer phenotype (theoretically freeing them from need for Q) were excluded from efficacy analyses. By change in CNS-LS score (defined as baseline score subtracted from the mean for days 15 and 29), DM/Q was significantly superior to its components, with a least-squares mean improvement, adjusted for baseline and center, of 7.4 points, compared with 4.1 for DM alone and 3.7 for Q alone. The average weekly PBA-episode rate during the study (from patient-diary data) was 1.9 times lower for DM/Q than for DM alone (excluding an outlier whose rate was tenfold greater than that of any other subject) and 2.1 times lower than for Q alone. During the final 2 weeks of the study, 52% of the DM/Q group had no PBA episodes, compared with 23% of the DM-monotherapy group and 12% of the Q-monotherapy group.
For a 12-week, randomized, double-blind study in MS [28], inclusion again required a clinical diagnosis of PBA and a baseline CNS-LS score ≥13. Depression was not assessed, except that major psychiatric disturbance was an exclusion criterion. During the study, 76 subjects received DM/Q 30/30 mg twice daily. The other 74 received placebo. Adjusted for baseline and center, the mean improvement in CNS-LS score (defined as baseline score subtracted from the mean for days 15, 29, 57, and 85) was significantly greater for DM/Q than for placebo, at 7.7 versus 3.3 points. Improvement in weekly PBA-episode rate was also significantly greater: in the DM/Q group, the mean decreased from 14.1 to 4.7, compared with a decrease from 17.3 to 11.5 in the placebo group. During weeks 9 through 12, 74% of DM/Q recipients had fewer than one episode per week, compared with 35% of placebo recipients.
In response to pharmacokinetic analyses suggesting that the Q dose in DM/Q could be lowered to 10 mg [35], the 12-week, randomized, double-blind Safety, Tolerability, And Efficacy Results (STAR; Clinicaltrials.gov #NCT00573443) study [27] was designed to have three treatment arms: DM/Q 30/10 mg, which 110 subjects received; DM/Q 20/10 mg, which 107 received; and placebo, which 109 received, once daily for the first week and twice daily thereafter. Subjects had a clinical diagnosis of PBA secondary to either ALS or MS and a CNS-LS score ≥13, and were excluded for significant depressive symptoms (Beck Depression Inventory version II [63] score >19) or a history of major psychiatric disturbance. The pre-specified primary efficacy analysis was of reduction in PBA-episode daily rate across the full study duration, as assessed by longitudinal negative binomial regression. For both DM/Q doses, the rate was significantly lower than for placebo, by 46.9% for DM/Q 30/10 mg and by 49.0% for DM/Q 20/10 mg. At endpoint, mean reduction in CNS-LS score was also significantly greater for both DM/Q doses than for placebo, at −8.2 points for 30/10 mg and −8.2 for 20/10 mg, compared with −5.7 for placebo. By responder analyses, the proportion of patients reporting PBA remission, defined as no episodes during the study’s final 2 weeks, was significantly greater for both DM/Q levels than for placebo, at 47.3% for 30/10 mg and 51.4% for 20/10 mg, compared with 29.4% for placebo. Although the STAR study yielded multiple signals favoring the 30/10 mg dose, including earlier emergence of significant improvement versus placebo in mean CNS-LS score and significant improvement versus placebo on the 36-Item Short-Form Health Survey [64] Mental Summary score and its social-functioning and mental-health subscores, both doses had similar efficacy on the primary and most secondary endpoints.
Patients completing the STAR study were eligible for a 12-week, open-label extension [65], during which all subjects took DM/Q 30/10 mg twice daily. CNS-LS scores continued to improve, by a mean 2.6 points for prior 30/10 mg, 2.4 points for prior 20/10 mg, and 3.1 points for prior placebo.
DM/Q Safety
In each of the studies of DM/Q for PBA, the active treatment generally had good safety and tolerability, with overall acceptable rates of expected adverse events (AEs). Among the AEs reported by ≥5% of DM/Q recipients in the STAR study (Table 2) [27], dizziness and diarrhea had a higher incidence in both of the DM/Q groups than in the placebo group, while nausea and urinary tract infection had highest incidence in the DM/Q 30/10 mg group, but rates in the DM/Q 20/10 mg group resembled those for placebo. Falls had a lower incidence in the DM/Q 20/10 mg group than in the DM/Q 30/10 mg group or the placebo group. Muscle spasms had highest incidence in the placebo group. The other AE types—headache, fatigue, somnolence, nasopharyngitis, constipation, muscle weakness, and dysphagia—had similar incidence in all groups (with differences of ≤2.6 percentage points across DM/Q 30/10 mg, DM/Q 20/10 mg, and placebo).
Informal comparisons of AE patterns across the three large-scale DM/Q trials suggest possible relationships of some AEs to the primary neurologic disease, the Q dosage (or its effect on DM levels), and the protocol for treatment initiation. In the 4-week ALS study [26], the frequency of discontinuation due to AEs was 24% for DM/Q 30/30 mg, compared with 6% for DM 30 mg and 5% for Q 30 mg, a difference potentially reflecting Q blockade of hepatic DM metabolism exclusively in the DM/Q group. Although the 12-week MS study [28] was three times longer than the ALS study, the frequency of discontinuation due to AEs was markedly lower, at 14.5% for DM/Q 30/30 mg and 10.8% for placebo, suggesting that DM/Q tolerability may vary by primary neurologic disease. In the 12-week STAR study [27], which used a smaller Q dose (10 mg) and a 1-week titration (with once-daily DM/Q dosing for the first week and twice-daily dosing thereafter, instead of twice-daily dosing from the start), AE-related discontinuation rates were even lower, at 5.5% for DM/Q 30/10 mg, 9.3% for DM/Q 20/10 mg, and 1.8% for placebo. In the STAR study, the frequency of nausea appeared to be DM dose-related, at 7.5% for DM/Q 20/10 mg and 12.7% for DM/Q 30/10 mg, which was markedly less than the 32.9% reported for DM/Q 30/30 mg in the 4-week ALS study [26] and the 22.4% reported for DM/Q 30/30 mg in the 12-week MS study [28]. Of interest, 9.2% of the STAR study’s placebo group also experienced nausea.
In the three studies, Q showed minimal QTc prolongation not deemed to be clinically relevant. However, the studies excluded patients with clinically significant cardiac disease or conduction abnormalities, and the DM/Q labeling includes cardiologic precautions and contraindications [25]. Special concern about respiratory function arises in ALS because DM is used as an over-the-counter antitussive, coughing is an important airway-clearing mechanism, and respiratory impairment is a prominent facet of ALS progression and mortality [66]. However, DM/Q showed no increased risk of adverse respiratory effects in any clinical trials. In the STAR study, for example, respiratory infections had a comparably low incidence in all treatment groups, no acute decompensations of respiratory function were observed, and oxygen-saturation data showed no clinically significant mean changes from baseline values [27].
Conclusion
PBA is a common, distressing, psychosocially disruptive dysregulation of emotional expression, occurring across a broad range of primary CNS disorders. Based on extensive clinical data showing efficacy, safety, and tolerability, DM/Q is the first pharmacotherapy specifically approved for treating PBA. Although antidepressants have been used to treat it, their utility has not been studied in well-controlled trials. Nor have any clinical studies either compared them with DM/Q or tested the outcomes of a switch in treatment. Comparing available studies of individual agents would be impeded by potential problems including the need to control for depression and for placebo effect.
Because of preclinical findings of DM effects on glutamatergic [29, 30, 32], serotonergic [31], and noradrenergic [31] neurotransmission as well as preliminary signals in clinical research [61, 67], DM/Q is being considered for other potential clinical applications. In December 2013, a Phase 2 study of its potential use against neuropathic pain in MS [68] was reported [69] to have failed to meet its primary efficacy outcome, pain reduction versus placebo [70]. DM/Q continues to be considered for potential use against neuropathic pain in other settings and against aggression or agitation in settings such as autism or dementia (Table 3). Meanwhile, expanded data may further characterize its efficacy and safety for PBA in settings other than ALS and MS. Expanded data may also illuminate long-term DM/Q use, which could last a patient’s lifetime but should be reassessed regularly. In PBA as in all other disorders, treatment decisions must always be appropriate for the individual patient.
References
Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and potential therapy. J Neuropsychiatry Clin Neurosci. 2005;17:447–54.
Miller A, Pratt H, Schiffer RB. Pseudobulbar affect: the spectrum of clinical presentation, etiologies and treatments. Expert Rev Neurother. 2011;11:1077–88.
Work S, Colamonico JA, Bradley WG, Kaye RE. Pseudobulbar affect: an under-recognized and undertreated neurological disorder. Adv Ther. 2011;28:586–601.
Siddiqui MS, Fernandez HH, Garvan CW, et al. Inappropriate crying and laughing in Parkinson disease and movement disorders. World J Biol Psychiatry. 2009;10:234–40.
Gallagher JP. Pathologic laughter and crying in ALS: a search for their origin. Acta Neurol Scand. 1989;80:114–7.
Robinson RG, Parikh RM, Lipsey JR, Starkstein SE, Price TR. Pathological laughing and crying following stroke: validation of a measurement scale and a double-blind treatment study. Am J Psychiatry. 1993;150:286–93.
Moore SR, Gresham LS, Bromberg MB, Kasarkis EJ, Smith RA. A self-report measure of affective lability. J Neurol Neurosurg Psychiatry. 1997;63:89–93.
Smith RA, Berg JE, Pope LE, Callahan JD, Wynn D, Thisted RA. Validation of the CNS emotional lability scale for pseudobulbar affect (pathological laughing and crying) in multiple sclerosis patients. Mult Scler. 2004;10:1–7.
Strowd RE, Cartwright MS, Okun MS, Haq I, Siddiqui MS. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257:1382–7.
Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29:775–98.
Lawson IR, MacLeod RD. The use of imipramine (“Tofranil”) and other psychotropic drugs in organic emotionalism. Br J Psychiatry. 1969;115:281–5.
Schiffer RB, Herndon RM, Rudick RA. Treatment of pathologic laughing and weeping with amitriptyline. N Engl J Med. 1985;312:1480–2.
Andersen G, Vestergaard K, Riis JO. Citalopram for post-stroke pathological crying. Lancet. 1993;342:837–9.
Brown KW, Sloan RL, Pentland B. Fluoxetine as a treatment for post-stroke emotionalism. Acta Psychiatr Scand. 1998;98:455–8.
Choi-Kwon S, Han SW, Kwon SU, Kang DW, Choi JM, Kim JS. Fluoxetine treatment in poststroke depression, emotional incontinence, and anger proneness: a double-blind, placebo-controlled study. Stroke. 2006;37:156–61.
Burns A, Russell E, Stratton-Powell H, Tyrell P, O’Neill P, Baldwin R. Sertraline in stroke-associated lability of mood. Int J Geriatr Psychiatry. 1999;14:681–5.
Murray V, von Arbin M, Bartfai A, et al. Double-blind comparison of sertraline and placebo in stroke patients with minor depression and less severe major depression. J Clin Psychiatry. 2005;66:708–16.
Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71:1193–207.
Parvizi J, Arciniegas DB, Bernardini GL, et al. Diagnosis and management of pathological laughter and crying. Mayo Clin Proc. 2006;81:1482–6.
Hackett ML, Yang M, Anderson CS, Horrocks JA, House A. Pharmaceutical interventions for emotionalism after stroke. Cochrane Database Syst Rev. 2010(2), Art. No. CD003690.
Udaka F, Yamao S, Nagata H, Nakamura S, Kameyama M. Pathologic laughing and crying treated with levodopa. Arch Neurol. 1984;41:1095–6.
Ramasubbu R. Lamotrigine treatment for post-stroke pathological laughing and crying. Clin Neuropharmacol. 2003;26:233–5.
Kishimoto A, Yatomi K, Yokoyama Y, Nakatsu N, Fujita K, Hashimoto K. Ifenprodil for emotional incontinence in patients with vascular dementia: two case reports. J Clin Psychopharmacol. 2013;33:143–5.
Kamurasaki Y, Yokoyama T, Ogura J, Maeda K. Treatment of pathologic emotionality with thyrotropin-releasing hormone. Jpn J Psychiatry Neurol. 1989;43:665–8.
NUEDEXTA® (dextromethorphan hydrobromide and quinidine sulfate) capsules [prescribing information]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc; October 2010.
Brooks BR, Thisted RA, Appel SH, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63:1364–70.
Pioro EP, Brooks BR, Cummings J, et al. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68:693–702.
Panitch HS, Thisted RA, Smith RA, et al. Randomized, controlled trial of dextromethorphan/quinidine for pseudobulbar affect in multiple sclerosis. Ann Neurol. 2006;59:780–7.
Choi DW, Peters S, Viseskul V. Dextrorphan and levorphanol selectively block N-methyl-d-aspartate receptor-mediated neurotoxicity on cortical neurons. J Pharmacol Exp Ther. 1987;242:713–20.
Musacchio JM, Klein M, Canoll PD. Dextromethorphan and sigma ligands: common sites but diverse effects. Life Sci. 1989;45:1721–32.
Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther. 1995;274:1263–70.
DeCoster MA, Klette KL, Knight ES, et al. Sigma receptor-mediated neuroprotection against glutamate toxicity in primary rat neuronal cultures. Brain Res. 1995;671:45–53.
Vetticaden SJ, Cabana BE, Prasad VK, et al. Phenotypic differences in dextromethorphan metabolism. Pharm Res. 1989;6:13–9.
Zhang Y, Britto MR, Valderhaug KL, et al. Dextromethorphan: enhancing its systemic availability by way of low-dose quinidine-mediated inhibition of cytochrome P4502D6. Clin Pharmacol Ther. 1992;51:647–55.
Pope LE, Khalil MH, Berg JE, et al. Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J Clin Pharmacol. 2004;44:1132–42.
Holford NH, Coates PE, Guentert TW, et al. The effect of quinidine and its metabolites on the electrocardiogram and systolic time intervals: concentration-effect relationships. Br J Clin Pharmacol. 1981;11:187–95.
Grace AA, Camm AJ. Quinidine. N Engl J Med. 1998;338:35–45.
Darwin C. The expression of the emotions in man and animals. London: John Murray; 1872.
Oppenheim H. Textbook of Nervous Diseases for Physicians and Students by Professor H. Oppenheim of Berlin. English translation by Alexander Bruce. London, T. N. Foulis Publisher; 1911.
Wilson SAK. Some problems in neurology II: Pathological laughing and crying. J Neurol Psychopathol. 1924;4:299–333.
Arciniegas DB, Lauterbach EC, Ginsburg DL, et al. The differential diagnosis of pseudobulbar affect (PBA): distinguishing PBA among disorders of mood and affect. CNS Spectr. 2005;10(suppl 5):1–16.
Cummings JL, Arciniegas DB, Brooks BR, et al. Defining and diagnosing involuntary emotional expression disorder. CNS Spectr. 2006;11:1–7.
Wortzel HS, Oster TJ, Anderson CA, Arciniegas DB. Pathological laughing and crying: epidemiology, pathophysiology and treatment. CNS Drugs. 2008;22:531–45.
Poeck K. Pathological laughing and weeping in patients with progressive bulbar palsy. Germ Med Mon. 1969;14:394–7.
Poeck K. Pathophysiology of emotional disorders associated with brain damage. In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology. New York: American Elsevier Publishing Company Inc; 1969. p. 343–67.
Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37:1893–916.
Allman P, Hope T, Fairburn GC. Crying following stroke. A report on 30 cases. Gen Hosp Psychiatry. 1992;14:315–21.
Olney NT, Goodkind MS, Lomen-Hoerth C, et al. Behavior, physiology and experience of pathological laughing and crying in amyotrophic lateral sclerosis. Brain. 2011;134:3458–69.
Parvizi J, Coburn KL, Shillcutt SD, Coffey CE, Lauterbach EC, Mendez MF. Neuroanatomy of pathological laughing and crying: a report of the American Neuropsychiatric Association Committee on Research. J Neuropsychiatry Clin Neurosci. 2009;21:75–87.
Okun MS, Raju DV, Walter BL, et al. Pseudobulbar crying induced by stimulation in the region of the subthalamic nucleus. J Neurol Neurosurg Psychiatry. 2004;75:921–3.
Sperli F, Spinelli L, Pollo C, Seeck M. Contralateral smile and laughter, but no mirth, induced by electrical stimulation of the cingulate cortex. Epilepsia. 2006;47:440–3.
Kahane P, Ryvlin P, Hoffmann D, Minotti L, Benabid AL. From hypothalamic hamartoma to cortex: what can be learnt from depth recordings and stimulation? Epileptic Disord. 2003;5:205–17.
Haiman G, Pratt H, Miller A. Brain responses to verbal stimuli among multiple sclerosis patients with pseudobulbar affect. J Neurol Sci. 2008;271:137–47.
Parvizi J, Anderson SW, Martin CO, Damasio H, Damasio AR. Pathological laughter and crying: a link to the cerebellum. Brain. 2001;124:1708–19.
Petty F, Davis LL, Kabel D, Kramer GL. Serotonin dysfunction disorders: a behavioral neurochemistry perspective. J Clin Psychiatry. 1996;57(Suppl 8):11–6.
Wroblewski JT, Danysz W. Modulation of glutamate receptors: molecular mechanisms and functional implications. Annu Rev Pharmacol Toxicol. 1989;29:441–74.
Askmark H, Aquilonius SM, Gillberg PG, et al. A pilot trial of dextromethorphan in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1993;56:197–200.
Blin O, Azulay JP, Desnuelle C, et al. A controlled one-year trial of dextromethorphan in amyotrophic lateral sclerosis. Clin Neuropharmacol. 1996;19:189–92.
Smith RA. Dextromethorphan/quinidine: a novel dextromethorphan product for the treatment of emotional lability. Expert Opin Pharmacother. 2006;7:2581–98.
Smith RA, Moore SR, Gresham LS, Manley PE, Licht JM. The treatment of affective lability with dextromethorphan [abstract 604P]. Neurology. 1995;45(Suppl 4):A330.
Smith RA, Licht JM, Pope LE, Berg JE, Arnold R. Combination dextromethorphan and quinidine in the treatment of frustration and anger in patients with involuntary emotional expression disorder (IEED) [abstract M-63]. Ann Neurol. 2006;60(S10):S50.
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62.
Beck AT, Steer RA, Brown GK. Manual for the Beck Depression Inventory–II. San Antonio (TX): Psychological Corporation; 1996.
Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30:473–83.
Pioro EP, Brooks BR, Cummings J, et al. Persistent efficacy of dextromethorphan/quinidine for pseudobulbar affect: results from a 12-week, open-label extension study [Poster P02.295]. Presented at: The 62nd Annual Meeting of the American Academy of Neurology; Toronto, April 10–17, 2010.
Kaplan LM, Hollander D. Respiratory dysfunction in amyotrophic lateral sclerosis. Clin Chest Med. 1994;15:675–81.
Wynn D, Schiffer R, Brooks BR, et al, on behalf of the STAR Trial investigators. Double-blind, placebo-controlled, multinational study of AVP-923 for pseudobulbar affect in MS [Poster A11]. In: Presented at: The 3rd World Congress on Controversies in Neurology (CONy); Prague, October 8–11, 2009.
Safety and efficacy of AVP-923 in the treatment of central neuropathic pain in multiple sclerosis. Available at: http://clinicaltrials.gov/show/NCT01324232. Accessed Feb 5, 2014.
Prior A. Avanir neuropathic pain treatment study did not meet primary efficacy endpoint; shares fall. Wall Street Journal, December 10, 2013. http://online.wsj.com/article/BT-CO-20131210-710884.html?dsk=y. Accessed Feb 5, 2014.
Farrar JT, Young JP, LaMoreaux L, Werth JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain. 2001;94:149–58.
Acknowledgments
This project was funded by Avanir Pharmaceuticals, Inc. Avanir was invited to provide comments for author consideration. However, the author had final control of the information presented and is solely responsible for its content. Any views expressed are those of the author. Editorial assistance in preparation of the manuscript was provided by Linnéa C. Elliott and Michael Feirtag of The Curry Rockefeller Group, LLC, Tarrytown, NY, USA. Support for this assistance was funded by Avanir Pharmaceuticals, Inc. The patient video was provided by John Fellus, MD, of The International Brain Research Foundation, Flanders, NJ USA. The named author meets the ICMJE criteria for authorship for this manuscript, takes responsibility for the integrity of the work as a whole, and has given final approval for the version to be published.
Conflict of interest
Erik P. Pioro has been a principal investigator of clinical trials, a member of scientific advisory board, and a speaker for Avanir Pharmaceuticals, Inc.
Compliance with ethics guidelines
This review article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by the author. Informed consent was obtained from the patient for the publication of the video (Video 1 in the electronic supplementary material).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (MP4 27311 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pioro, E.P. Review of Dextromethorphan 20 mg/Quinidine 10 mg (NUEDEXTA®) for Pseudobulbar Affect. Neurol Ther 3, 15–28 (2014). https://doi.org/10.1007/s40120-014-0018-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-014-0018-5