
Abstract
Background
Platelet inhibition is crucial in reducing both short- and long-term atherothrombotic risks in patients with acute coronary syndromes (ACS) managed with percutaneous coronary intervention (PCI). Based on randomised trials, recent recommendations in the current guidelines include the endorsement of prasugrel as a first-choice adenosine diphosphate receptor inhibitor. Yet, there is limited experience with the use of prasugrel in routine practice.
Methods
The Rijnmond Collective Cardiology Research (CCR) registry is a prospective, observational study that will follow-up 4000 PCI-treated ACS patients in the larger region of Rotterdam, the Netherlands. Based on recently implemented hospital protocols, all patients will receive prasugrel as first-choice antiplatelet agent, unless contraindicated, in accordance with European guidelines, and will be followed for up to 1 year post-discharge for longitudinal assessment of outcomes and bleeding events. This registry exemplifies a collaborative study design that employs a regional PCI registry platform and provides feedback to participating sites regarding their practice patterns, thereby supporting and promoting improvement of quality of care.
Conclusion
The CCR registry will evaluate the adoption of prasugrel into routine clinical practice and thus, will provide important evidence with regard to the benefits and risks of real-world utilisation of prasugrel as antiplatelet therapy in PCI-treated ACS patients.
Similar content being viewed by others

Background
Platelet inhibition is crucial for reducing the short- and long-term risks in patients with acute coronary syndromes (ACS) [1–3]. Therefore, dual antiplatelet therapy with aspirin and a thienopyridine for the prevention of thrombotic complications is recommended in practice guidelines and represents the standard therapy, particularly in those undergoing percutaneous coronary intervention (PCI). Although clopidogrel has been the most widely used adenosine diphosphate (ADP) receptor inhibitor to date, pharmacodynamic interactions and genetic polymorphisms result in significant variability in the antiplatelet effect of clopidogrel [4–6], leading to the concern that some patients may be at increased risk for thrombotic events [7–9]. Such limitations prompted the search for alternative ADP receptor inhibitors that may provide more potent and more consistent platelet inhibition.
Prasugrel is a third-generation thienopyridine prodrug that is more efficiently metabolised to its active metabolite than clopidogrel and demonstrates a faster onset of action with a more consistent and greater degree of platelet inhibition [10]. Based on the results of the TRial to assess Improvement in Therapeutic Outcomes by optimising platelet inhibitioN with prasugrel—Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 [11], prasugrel was approved in Europe as well as the United States. This study randomised 13,608 patients with moderate-to-high risk ACS scheduled for PCI to prasugrel (60 mg loading dose, 10 mg maintenance dose) or clopidogrel (300 mg loading dose, 75 mg maintenance dose). Prasugrel treatment was associated with a significant 19 % relative risk reduction (RR) (hazard ratio 0.81, 95 % confidence interval [CI] 0.73–0.90; p < 0.001) and a 2.2 % absolute RR (9.9 % vs. 12.1 %; p < 0.001) of the primary efficacy endpoint (composite of cardiovascular mortality, nonfatal myocardial infarction, or nonfatal stroke), along with other ischaemic events compared with clopidogrel. However, the increased efficacy of prasugrel was counterbalanced by a clinically significantly increased risk of bleeding, particularly among patients with a history of stroke or transient ischaemic attack (TIA), as well as elderly (≥75 years) or underweight patients (<60 kg) [11].
Based on the results of the TRITON-TIMI 38 trial [11] and updated guidelines of the European Society of Cardiology (ESC) for non-ST-segment elevation acute coronary syndromes (NSTE-ACS) [12] and myocardial revascularisation [13], as well as updated guidelines of the American College of Cardiology/American Heart Association (ACC/AHA) [14], in 2011, our network instituted prasugrel as first-choice treatment option in patients with ACS undergoing PCI. As this represents a major change in the treatment of these patients, we sought to monitor the implementation and clinical outcomes by means of an observational study. Observational studies can confirm the generalisability of randomised clinical trial findings among a broader spectrum of patients, and allow investigation of comparative effectiveness and safety of antiplatelet regimens in real-world settings [15]. The Rijnmond Collective Cardiology Research (CCR) study is a clinical registry that will provide comprehensive longitudinal assessment of post-ACS treatment patterns and outcomes in routine clinical practice. In this article, we describe the design of the CCR study.
Methods
Study design and population
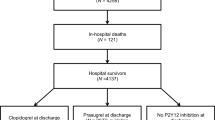
The CCR study (Dutch Trial Register unique identifier: NTR3704) is a prospective observational longitudinal registry, which will be conducted at 11 member sites of the CCR network in the Rijnmond region in the Netherlands. Prospective sites will include 8 referring hospitals and 3 interventional centres which are listed in Appendix A. The CCR study will enrol up to 4000 patients with ACS who are undergoing PCI during the index hospitalisation and who are treated with an ADP receptor inhibitor (Fig. 1). Prior to enrolment of the first patient on 1 August 2011, treatment guidelines of the participating network were updated to include prasugrel as a first-line treatment option for antiplatelet therapy, based on the available ESC and the ACC/AHA guideline recommendations at that time. In essence, all patients receive prasugrel, unless contraindicated. There is no treatment intervention directed by the protocol of the CCR study. All treatment decisions are at the discretion of the individual treating physician in accordance with practice guideline recommendations and local standards of care and practice. The broad eligibility criteria are outlined in Table 1.

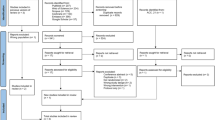
Longitudinal study design of the CCR study. *Clopidogrel 600 mg loading dose and 75 mg/daily maintenance dose when prasugrel is contraindicated. ACS indicates acute coronary syndrome; CABG coronary artery bypass grafting; CV cardiovascular; LD loading dose; MACE major adverse cardiac events; MD maintenance dose; MI myocardial infarction; PCI percutaneous coronary intervention; ST stent thrombosis; and TVR target vessel revascularisation
Ethics
For the purpose of this study patients will not be subjected to acts or be imposed to any mode of behaviour, otherwise than their regular treatment. Therefore, according to Dutch law, written informed consent for a patient to be enrolled in this study is not required. This study is conducted according to the Privacy Policy of the Erasmus MC and according to the Erasmus MC regulations for the appropriate use of data in patient-oriented research, and is approved by the regional ethics committee (reference # MEC-2010-417).
Study objectives and endpoints
The primary objective of the CCR study is to monitor clinically relevant parameters of ACS patients undergoing PCI with a follow-up of 1 year. The parameters will include baseline patient characteristics, drug treatments (including timings and logistic parameters), and clinical outcomes. Other key secondary objectives are detailed in Table 2. The primary endpoint of the study is a composite of all-cause mortality and nonfatal myocardial infarction. Definitions of the other effectiveness and safety endpoints are provided in Table 3.
Data collection, data management and event validation
Patient characteristics, clinical features, angiographic and procedural details, and in-hospital outcomes are abstracted from the medical chart per routine and entered into a secure web-based and centralised database by enrolling site personnel. Data quality is maintained by various quality measures, including integrated data check systems with plausibility thresholds. All collected data elements are subject to additional data checks with queries to enrolling sites, thereby maximising completeness of data and quality. After the index hospitalisation, patients are routinely followed up at 1 month (±2 week) and 12 months (+2 months) at the outpatient clinics of the enrolling sites, after which longitudinal information on patient treatment, effectiveness and safety outcomes is collected and entered into the central database. Independent data monitoring on-site at each enrolling centre is conducted monthly with feedback and training of site data managers.
When study endpoints are suspected, this will be entered into the central database by site personnel. Hereafter, additional documents, including relevant hospital discharge summaries, procedural reports, or angiographic films, will be obtained for event validation by an independent clinical endpoint committee.
Sample size determination
Precision in measurement and estimation corresponds to the reduction of random error; it can be improved by increasing the size of the study. Approximately 4000 patients will be enrolled in the CCR study. Previous real-world data in our region showed an overall 1-year rate of at least 8.0 % for the primary combined endpoint (composite of all-cause mortality and myocardial infarction) in PCI-treated ST-segment elevation myocardial infarction (STEMI) patients. This corresponds with a 95 % CI of 8.0 ± 0.8 % for a population of 4000 patients and, thus, provides a statistically precise estimate in the current study. In NSTE-ACS patients undergoing PCI, we expect an overall 1-year rate of 5.0 %, corresponding with a 95 % CI extending from 5.0 % ± 0.7 % in a dataset containing 4000 patients. For subgroup analyses including 1000 observations, the 95 % CI will be 5.0 % ± 1.4 %.
Statistical analysis
Continuous variables will be summarised as mean ± standard deviation or median with interquartile range, depending on normal distribution and will be compared using the Student t-test or Mann–Whitney U-test as appropriate. Categorical variables will be summarised as proportions and compared using the chi-square test. Whenever necessary, multivariable analyses, such as binary logistic regression analyses, will be performed. Differences between groups in time-to-event endpoints will be assessed with the log-rank test. Survival probabilities will be displayed and calculated using Kaplan-Meier methodology. Hazard ratios between groups will be calculated using Cox proportional hazard model. Two-sided P-values <0.05 will be considered statistically significant.
Study cohorts will be determined based on the ADP receptor inhibitor received at the time of the index PCI. For patients who discontinue or change to a different ADP receptor inhibitor, events that occur within 7 days of medication switch or discontinuation will be counted toward the ADP receptor inhibitor at the time of index PCI. Events that occur >7 days after the medication switch or discontinuation will be censored in the primary analysis.
For determination of real-world comparative benefits of prasugrel relative to clopidogrel, as outlined in the secondary objectives (Table 2), we will use existing outcome data in the Erasmus MC data repository of historical clopidogrel-treated patients.
Study organisation
The CCR study is supported by Eli-Lilly Nederland B.V. and Daiichi Sankyo Nederland B.V. Other than providing financial support, they are not involved in the study design, study management, or data interpretation. The CCR steering committee, composed of experienced clinical investigators in interventional cardiology (Appendix B), is solely responsible for the design and conduct of this study, all study analyses, and the drafting and editing of this and forthcoming manuscripts. The steering committee will have full access to the final study data.
An independent clinical endpoint committee will adjudicate efficacy endpoint events as well as bleeding events. As the CCR registry is an observational study, there are no provisions for a data safety monitoring board. Enrolling sites and treating physicians will follow all applicable Dutch laws and regulations for reporting events to authorities and/or local market authorisation holder.
Timeline
The first patient was enrolled on 1 August 2011. The anticipated duration of enrolment is approximately 18 months or until the intended number of 4000 patients is recruited.
Discussion
The CCR registry is a prospective, multicentre, longitudinal, observational study designed to assess the in-hospital and 1-year care patterns and outcomes in ACS patients treated with PCI and antiplatelet therapy. As our network instituted prasugrel as the first-line treatment option in patients with ACS undergoing PCI, this study provides an ideal means of evaluating the adoption of this novel agent in routine practice. Recommendations from the ESC guidelines stress the importance of the development of regional and/or national programs to measure performance indicators systematically that can provide feedback to individual hospitals [12]. In addition, the participation in standardised quality-of-care data registry for tracking and measuring outcomes and complications, among other goals, is encouraged in the ACC/AHA guidelines [14]. Not only will the CCR study allow the comparison of prasugrel with historical clopidogrel-treated patients with respect to clinical outcomes, and thereby confirm the generalisability of randomised findings among a broader spectrum of patients, but also important evidence will be provided concerning several unanswered questions with regard to the use of prasugrel in routine practice, which are discussed below.
Pretreatment with prasugrel
Based on current guidelines, the use of prasugrel in NSTE-ACS patients should be restricted to patients with known coronary anatomy [12]. On the other hand, these guidelines also recommend the administration of a thienopyridine as soon as possible when ACS is suspected [12] and state that it is not desirable to withhold optimal medical therapy until intervention. Indeed, the advantage of early administration of a thienopyridine loading dose for patients with unknown coronary anatomy prior to diagnostic angiography would be to achieve higher inhibition of platelet aggregation and thus prevent recurrent atherothrombotic events in patients likely to undergo PCI. Therefore, in our network, prasugrel is already initiated upstream in the ambulance during transportation for primary PCI in STEMI patients. Randomised clinical trials in which the effect of thienopyridine pretreatment (timings) in ACS is being investigated are ongoing. The ACCOAST trial will compare two prasugrel loading dose schedules in patients with NSTEMI who are scheduled for coronary angiography/PCI [16]. The effect of an early pretreatment (30 mg loading dose after diagnosis followed by coronary angiography with an additional dose of 30 mg given at the time of PCI) on clinical outcome will be compared with non-pretreatment (standard 60 mg loading dose at the time of PCI) [16]. The ATLANTIC trial investigates prehospital vs. in-hospital use of ticagrelor in patients with STEMI referred for primary PCI (ClinicalTrials.gov identifier: NCT01347580). In this regard, the CCR study will provide real-world data concerning the timing of pretreatment with prasugrel and outcomes in patients with ACS. Patients will receive a 60 mg loading dose of prasugrel before arrival at the cathlab either prehospital (ambulance), in the referring hospital or in-hospital (emergency room or coronary care unit). Our observations will extend the findings from randomised trials and will provide important evidence from real-world practice.
Bleeding risk and subgroups
As expected, the improved efficacy of prasugrel in the TRITON-TIMI 38 trial was associated with higher rates of bleeding [11]. Particularly, the elderly (≥75 years) and underweight patients (<60 kg) tended to bleed more frequently with the conventional dose (10 mg) of prasugrel. While in the elderly and underweight patients the net clinical benefit (primary endpoint plus major haemorrhage) of prasugrel was comparable with clopidogrel, patients with a history of stroke or TIA were at higher risk for bleeding with unfavourable outcome [11]. Based on these findings, prasugrel is contraindicated in patients with previous stroke or TIA. However, despite the lack of clinical outcome data as well as limited pharmacodynamic data [17], a lower maintenance dose (5 mg instead of 10 mg) is recommended by the European Medicines Agency for elderly and/or underweight patients. Whether the proposed reduction of the maintenance dose of prasugrel to 5 mg is efficacious and safe will also be investigated in the CCR study.
Triple therapy
The management of patients on oral anticoagulation (e.g. for atrial fibrillation, mechanical heart valves) requiring antiplatelet therapy after PCI remains another challenging issue. Since there are limited data available, both the ESC and the ACC/AHA have marginally included recommendations in the guidelines regarding triple therapy. Different clinical scores concerning thrombotic (CHA2DS2-VASC score) [18, 19] and bleeding (HAS-BLED score) [20] risk may provide additional guidance in therapeutic decisions in such patients. Importantly, however, the CCR study will provide real-world data regarding treatment patterns and bleeding outcomes in patients prone to triple therapy.
Ticagrelor
Along with prasugrel, the use of ticagrelor as antiplatelet therapy in ACS patients undergoing PCI is also recommended in current guidelines [12, 13, 21]. Ticagrelor, a non-thienopyridine ADP receptor inhibitor causing reversible inhibition of platelet aggregation, has been tested against clopidogrel in the Study of Platelet Inhibition and Patient Outcomes and proved beneficial with respect to a combined clinical outcome including mortality [22]. The rate of severe non-CABG-related bleeding [22] was similar to that of prasugrel in the TRITON-TIMI 38 trial [11], whereas CABG-related bleeding was lower than for clopidogrel [22]. In patients with STEMI, ticagrelor lost its statistically significant superiority over clopidogrel with respect to the primary combined endpoint (10.8 % vs. 9.4 %; 13 % relative RR; p = 0.07) [23], whereas a pronounced benefit was demonstrated in a STEMI population when treated with prasugrel (12.4 % vs 10.0 %; 21 % relative RR; p = 0.02) [24]. However, as there are no head-to-head comparison studies of prasugrel and ticagrelor, potential differences between the two agents remain hypothetical.
Conclusion
The CCR registry is a prospective, longitudinal, observational study designed to evaluate the adoption of prasugrel into routine clinical practice. This study will provide important evidence regarding the benefits and risks of the use of prasugrel as initial antiplatelet treatment strategy in patients with ACS undergoing PCI. Along with providing comparative effectiveness with clopidogrel-treated patients, and thereby extending randomised findings across a broad array of patient types and practice settings, crucial real-world data, including pretreatment, bleeding risk in certain subgroups, and triple therapy will be gathered in the near future with regard to the use of prasugrel in real-world situations. Finally, the CCR study is in accordance with guidelines stressing the importance of quality improvement and performance assessment in the management of ACS patients.
References
Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358(9281):527–33.
Patrono C, Bachmann F, Baigent C, et al. Expert consensus document on the use of antiplatelet agents. The task force on the use of antiplatelet agents in patients with atherosclerotic cardiovascular disease of the European society of cardiology. Eur Heart J. 2004;25(2):166–81.
Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494–502.
Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360(4):354–62.
Serebruany VL, Steinhubl SR, Berger PB, et al. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol. 2005;45(2):246–51.
Rademakers LM, Dewilde W, van de Kerkhof D. Early double stent thrombosis associated with clopidogrel hyporesponsivenesss. Neth Heart J. 2012;20(1):38–41.
Barragan P, Bouvier JL, Roquebert PO, et al. Resistance to thienopyridines: clinical detection of coronary stent thrombosis by monitoring of vasodilator-stimulated phosphoprotein phosphorylation. Catheter Cardiovasc Interv. 2003;59(3):295–302.
Matetzky S, Shenkman B, Guetta V, et al. Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation. 2004;109(25):3171–5.
Wiviott SD, Antman EM. Clopidogrel resistance: a new chapter in a fast-moving story. Circulation. 2004;109(25):3064–7.
Jakubowski JA, Winters KJ, Naganuma H, et al. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007;25(4):357–74.
Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–15.
Hamm CW, Bassand JP, Agewall S, et al. ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2011;32(23):2999–3054.
Wijns W, Kolh P, Danchin N, et al. Guidelines on myocardial revascularization. Eur Heart J. 2010;31(20):2501–55.
Wright RS, Anderson JL, Adams CD, et al. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American Academy of Family Physicians, Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;57(19):e215–367.
Shah BR, Drozda J, Peterson ED. Leveraging observational registries to inform comparative effectiveness research. Am Heart J. 2010;160(1):8–15.
Montalescot G, Bolognese L, Dudek D, et al. A comparison of prasugrel at the time of percutaneous coronary intervention or as pretreatment at the time of diagnosis in patients with non-ST-segment elevation myocardial infarction: design and rationale for the ACCOAST study. Am Heart J. 2011;161(4):650–6 e1.
Breet NJ, van Donkersgoed HE, van Werkum JW, et al. Is platelet inhibition due to thienopyridines increased in elderly patients, in patients with previous stroke and patients with low body weight as a possible explanation of an increased bleeding risk? Neth Heart J. 2011;19(6):279–84.
Lip GY. Anticoagulation therapy and the risk of stroke in patients with atrial fibrillation at ‘moderate risk’ [CHADS2 score = 1]: simplifying stroke risk assessment and thromboprophylaxis in real-life clinical practice. Thromb Haemost. 2010;103(4):683–5.
Lip GY, Nieuwlaat R, Pisters R, et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the euro heart survey on atrial fibrillation. Chest. 2010;137(2):263–72.
Lip GY, Frison L, Halperin JL, et al. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: the HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol Concomitantly) score. J Am Coll Cardiol. 2011;57(2):173–80.
Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/Non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2012;126(7):875–910.
Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–57.
Steg PG, James S, Harrington RA, et al. Ticagrelor versus clopidogrel in patients with ST-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: a Platelet Inhibition and Patient Outcomes (PLATO) trial subgroup analysis. Circulation. 2010;122(21):2131–41.
Montalescot G, Wiviott SD, Braunwald E, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): double-blind, randomised controlled trial. Lancet. 2009;373(9665):723–31.
Acknowledgements
The authors would like to thank Elisabeth Huijskens for her excellent contribution to the monitoring process. The authors would also like to acknowledge the professionalism and commitment to this study of the staff at all interventional and referring hospitals participating in the CCR collaboration.
Conflict of interest
Financial support to the CCR study is provided by the Eli-Lilly Nederland B.V. and Daiichi Sankyo Nederland B.V.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Member hospitals are listed in Appendix A
Appendices
Appendix A
Participating centres
Albert Schweitzer Hospital, Dordrecht; Beatrix Hospital, Gorinchem; Erasmus MC, Rotterdam; Havenziekenhuis, Rotterdam; IJsselland Hospital, Rotterdam; Ikazia Hospital, Rotterdam; Maasstad Hospital, Rotterdam; Ruwaard van Putten Hospital, Spijkenisse; Sint Franciscus Gasthuis, Rotterdam; Van Weel-Bethesda Hospital, Dirksland; and Vlietland Hospital, Schiedam. All centres are in the Netherlands.
Appendix B
Steering committee
A.G. de Vries, MD, Albert Schweitzer Hospital, Dordrecht; E. Boersma, PhD, Erasmus MC, Rotterdam; M.M.J.M. van der Linden, MD, PhD, Vlietland Hospital, Schiedam (principal investigator); P.C. Smits, MD, PhD, Maasstad Hospital, Rotterdam; and R.J.M. van Geuns, MD, PhD, Erasmus MC, Rotterdam.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Yetgin, T., van der Linden, M.M.J.M., de Vries, A.G. et al. Adoption of prasugrel into routine practice: rationale and design of the Rijnmond Collective Cardiology Research (CCR) study in percutaneous coronary intervention for acute coronary syndromes. Neth Heart J 22, 55–61 (2014). https://doi.org/10.1007/s12471-013-0472-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-013-0472-1