
Abstract
Relatively little is known about how genetic influences on alcohol abuse and dependence (AAD) change with age. We examined the change in influence of genetic and environmental factors which explain symptoms of AAD from adolescence into early adulthood. Symptoms of AAD were assessed using the four AAD screening questions of the CAGE inventory. Data were obtained up to six times by self-report questionnaires for 8,398 twins from the Netherlands Twin Register aged between 15 and 32 years. Longitudinal genetic simplex modeling was performed with Mx. Results showed that shared environmental influences were present for age 15–17 (57%) and age 18–20 (18%). Unique environmental influences gained importance over time, contributing 15% of the variance at age 15–17 and 48% at age 30–32. At younger ages, unique environmental influences were largely age-specific, while at later ages, age-specific influences became less important. Genetic influences on AAD symptoms over age could be accounted for by one factor, with the relative influence of this factor differing across ages. Genetic influences increased from 28% at age 15–17 to 58% at age 21–23 and remained high in magnitude thereafter. These results are in line with a developmentally stable hypothesis that predicts that a single set of genetic risk factors acts on symptoms of AAD from adolescence into young adulthood.
Similar content being viewed by others

Introduction
Twin and adoption studies have consistently shown that the risk for a lifetime history of alcohol abuse/dependence (AAD) is strongly influenced by genetic factors with estimates of heritability ranging around 28% in adolescents (Dick et al. 2009) and from 45 to 64% in adults (Cloninger et al. 1981; Goodwin et al. 1973; Heath et al. 1997; Kendler et al. 1992, 1997; Knopik et al. 2004; Liu et al. 2004; Pickens et al. 1991; Prescott and Kendler 1999; Reed et al. 1996; Sartor et al. 2010; True et al. 1999). These cross-sectional studies could however not examine whether AAD in adolescence and adulthood is influenced by the same or different genetic factors. For this, longitudinal studies are needed. Within a longitudinal design, the relative contributions of genetic and environmental influences have been explored from adolescence into young adulthood for a wide range of alcohol-related phenotypes. Generally, with increasing age the importance of additive genetic factors increases and that of shared environmental influences decreases for alcohol use measures such as alcohol initiation (Koopmans and Boomsma 1996; Viken et al. 1999), frequency of alcohol use (Geels et al. 2011; Rose et al. 2001; Viken et al. 1999), quantity of alcohol use (Kendler et al. 2008c; Geels et al. 2011) and perceived alcohol availability (Gillespie et al. 2007). In contrast to alcohol use, the change of the genetic underpinnings of symptoms of alcohol misuse during adolescence and early adulthood, has seldomly been explored in a longitudinal design.
In this article, the primary goal is to investigate how genetic risk factors for symptoms of AAD change across age. In particular, we seek to discriminate between two hypotheses about the developmental pattern of genetic risk factors for AAD symptoms in the key time period of adolescence and early adulthood. In this period drinking habits are commonly formed, levels of alcohol consumption typically peak (Koppes et al. 2000; Midanik and Clark 1994; Moore et al. 2005; Poelen et al. 2005) and symptoms of AAD usually begin (Harford et al. 2005; Schuckit et al. 1998). The developmentally stable hypothesis predicts that a single set of genetic risk factors will impact on AAD symptoms from late adolescence through early adulthood. By contrast, the developmentally dynamic hypothesis predicts that new genetic influences on AAD symptoms “come on line” at a particular age. These genetic innovations give rise to a qualitative change in genetic effects. Regardless of qualitative change, the importance of genetic risk factors may change quantitatively over time. This can result from genetic amplification, if the importance of genetic influences increases over age, or from genetic attenuation if the importance of genetic factors declines during development.
The current study analyzed longitudinal survey data on symptoms of AAD in a cohort of 8,398 twins from the Netherlands Twin Register aged between 15 and 32 years. Symptoms of AAD were assessed by the CAGE, a four item screening instrument for AAD, and were obtained up to six times. To ensure data from participants of different ages and sex could be meaningfully compared, measurement invariance of the CAGE was tested over age and sex. In addition to the primary goal of clarifying the nature of genetic effects on AAD symptoms over age, we also examined qualitative and quantitative changes of environmental risk factors. To achieve this aim, simplex models were fitted by which genetic and environmental influences could be separated from variance due to measurement error and the presence of genetic and environmental innovation could be tested.
Methods
Sample
Data came from a sample of twins (aged 15–32 years; N = 8,398) who were registered with the Netherlands Twin Register (NTR) and had answered survey questions about symptoms of AAD as part of the ongoing longitudinal study on health, personality and lifestyle that is being conducted in all active participants (twins and their relatives) of the Adult Netherlands Twin Register (ANTR) since 1991 (Boomsma et al. 2002, 2006). Questions about AAD symptoms were included in the surveys in 1995, 1997, 2000, 2002, 2004/2005 and 2009/2010. In 2009/2010, twins of 18 years and older (and their family members) who were followed since their birth by the Young Netherlands Twin Register (YNTR) (Bartels et al. 2007) were also invited to take part in the ANTR study. Extended information about the NTR sample and data collection can be found in Boomsma et al. (2002, 2006).
In our sample, very few participants under the age of 15 reported symptoms of AAD (see also Poelen et al. 2005). Therefore, to study the genetic architecture of AAD symptoms during adolescence and early adulthood, we included all participants aged 15 or older with data on symptoms of AAD. Because we collected data on symptoms of AAD from 1995 to 2010, spanning a 16 year period, the upper age limit was 32 years. Thus, for this study data on symptoms of AAD were analyzed if obtained between the ages 15 and 32. Zygosity for same-sex twin pairs was based on DNA polymorphisms if available (42%), or otherwise on survey questions about zygosity (58%). Agreement between DNA zygosity and zygosity based on survey questions for same-sex twins was 97% (Willemsen et al. 2005). The 8,398 participants (62% female; year of birth 1964–1991) provided up to six measurements of AAD symptoms. Amongst this group, 1.9% had reported that they had not or rarely drunk alcohol. For 1,588 individuals (18.9%) two measurements on AAD symptoms were analyzed, for 1,175 (14.0%) three, for 778 (9.3%) four, for 344 (4.1%) five and for 24 individuals (0.3%) six measurements. Additional repeated measures at age 14 or younger or age 33 or older were available but not analyzed for 674 of the 8,398 individuals (8.0%). The relative large number of twins with one survey (N = 4,489; 53.5%) was mainly due to the entry from participants of the YNTR in 2009/2010 who were invited to participate in the ANTR research for the first time (N = 2,151; 47.9%).
Measure
Symptoms of AAD were assessed by the CAGE, originally developed as a screening instrument for medical settings (Ewing 1984), which gives an indication of the presence of symptoms of AAD (Dhalla and Kopec 2007). The CAGE consists of four items that can be answered with ‘yes’ or ‘no’: Have you ever felt that you should Cut down your drinking?; Have people Annoyed you by criticizing your drinking?; Have you ever felt bad or Guilty about your drinking?; Have you ever had a drink in the morning (as an ‘Eye opener’) to steady your nerves or get rid of a hangover? In 2009/2010, there were three response categories: ‘yes, during the last year’; ‘yes, not during the last year’. These two yes categories were recoded into one, since its frequency was comparable to the frequency of the single yes categories in earlier years. If one of the four CAGE items was missing (N = 64; 0.8%), its value was imputed based on the mean of the three other answers. In case of two or more missing answers (N = 30; 0.4%), the CAGE was set to missing.
The number of participants aged 15–17 who gave two or more positive responses on the CAGE was low (see Table 1). This leads to numerical problems when analyzing longitudinal twin data. Therefore, the analyses were conducted on the dichotomy of zero vs. one or more yes answers. In analyses that are described in more detail below, we investigated whether this dichotomization influenced the results.
Data file restructuring
The mean age across surveys was 19.7 (SD 2.9) in 1995, 21.6 (SD 3.6) in 1997, 24.2 (SD 3.9) in 2000, 26.4 (SD 3.7) in 2002, and 26.9 (SD 3.6) in 2004/2005. Due to the entry of a large new group of young participants, the mean age was 22.9 (SD 4.9) in 2009/2010. To study age-related changes in genetic influences on AAD symptoms, the data file was restructured from observations by survey to observations by age band. Six age groups were created: 15–17, 18–20, 21–23, 24–26, 27–29, and 30–32, since surveys were sent out every 2–3 years. If two or more observations of one individual fell into the same age band, the most recent measurement was selected (at which the co-twin also answered the CAGE). Table 2 shows the number of observations for each age group, separately for sex and zygosity.
Data analysis
Data analysis consisted of four steps: testing whether the CAGE was measurement invariant over age and sex, estimation of longitudinal and twin correlations, cross-sectional genetic modeling and longitudinal genetic modeling. In all analyses, the CAGE was treated as a threshold character assuming that a standard normal liability distribution underlies the ordered categories.
Measurement invariance
We first investigated whether the CAGE was measurement invariant over age and sex. From the dataset as described above one observation per individual was selected. For individuals with repeated measures, observations were selected that gave age groups of roughly the same size. This resulted in the following 12 mutually exclusive age by sex groups: twins aged 15–17 (383 males, 691 females); twins aged 18–20 (698 males, 1,142 females); twins aged 21–23 (530 males, 754 females); twins aged 24–26 (353 males, 705 females); twins aged 27–29 (870 males, 1,487 females); twins aged 30–32 (343 males, 442 females).
In Mplus 5.1 (Muthén and Muthén 2007) one factor models were specified with the four CAGE items as indicators. First, a configural invariance model was fitted. In this model the factor loadings and thresholds were estimated for each group. Strong factorial invariance and strict factorial invariance models were tested in line with Slof-Op ‘t Landt et al. (2009). In the strong factorial invariance model, the factor loadings and thresholds were constrained to be equal over the 12 groups. The residual variances and means and variances of the latent factor were then estimated. In the strict factorial invariance model, the residual variances were also constrained to be equal over groups. This model tested if for all groups the same proportion of variance is explained by the latent factor. In an additional model, sex differences at the latent factor level were evaluated by constraining the factor means for males and females to be equal. Absence of sex differences at the latent factor level is not a requirement for measurement invariance, but makes the interpretation of results from the cross-sectional and longitudinal analyses easier. Model fit was evaluated with the RMSEA, a measure that indicates the approximate fit of the measurement model in the population, since the χ2 difference test is directly affected by sample size. An estimate in between .00 and .05 indicates good model fit (Schermelleh-Engel and Moosbrugger 2003). To correct for the non-independence of twin data, for all models option complex was used. This method was shown to perform satisfactory in the context of family data by Rebollo et al. (2006).
Estimation of longitudinal and twin correlations
To get a first impression of the relative importance of genetic and environmental influences on AAD symptoms, tetrachoric twin correlations for the dichotomous CAGE scores (0 vs. 1+ yes answers) were estimated. These analyses were performed with the package polycor (polychor(var1, var2, ML = T, std. err = T)) in R (version 2.9.2; http://www.r-project.org/) (R Development Core Team 2010) using maximum likelihood estimation. For each of the five zygosity by sex groups, six cross twin-within age correlations (30 in total), 15 cross twin-cross age correlations (75 in total), and 15 within person-cross age correlations were computed.
Sex differences in the correlations and the prevalence of one or more yes answers were tested cross-sectionally by model fitting in Mx (version 3.2) (Neale et al. 2006). In a multigroup analysis of five zygosity by sex groups, qualitative and quantitative sex differences in the correlations were tested for by constraining the correlations to be equal over sex. To examine sex differences in the prevalence, a sex effect on the threshold was tested for significance. The fit of the restricted models was compared to that of the full model using the likelihood ratio test.
To investigate whether the way the CAGE was dichotomized affected the results, correlations based on the dichotomous CAGE were compared to those based on the CAGE with three response categories: 0, 1, and 2+ yes answers. The polychoric correlations for the CAGE with three response categories were estimated for the age groups 18–20, 21–23, 24–26, 27–29 and 30–32 cross-sectionally, using Mx. For the 15–17 year olds the number of 2+ yes answers was too low to estimate polychoric correlations. In the analyses described above on a few occasions empty cells were observed (0.7–1.7%). These were replaced by 1 to be able to run the model estimation.
Genetic analyses
The relative importance of genetic and environmental influences on the CAGE was investigated by decomposing the variance–covariance structure of the dichotomous CAGE (0 vs. 1+ yes answers) into latent factors representing additive genetic (A), non-additive genetic (D) or shared environmental influences (C) and unique environmental (E) influences. First, cross-sectional genetic analyses were conducted for each age group separately. Next, longitudinal genetic analyses were performed. All genetic analyses were conducted in Mx (Neale et al. 2006) in two zygosity groups, as sex differences in the twin correlations were absent. For model identification purposes, the variance was fixed at unity for all age groups in both the cross-sectional models as well as in the longitudinal model.
Cross-sectional genetic analyses
The decision to fit an ACE or ADE model was based on the correlation structure for a particular age group. If the correlation for MZ twins was smaller than twice the correlation for DZ twins, an ACE model was fitted. If the MZ correlation was more than twice the DZ correlation, the ADE model was fitted to the data. The importance of additive genetic, non-additive genetic and shared environmental factors was tested by dropping the a, d or c parameter from the model and comparing the fit of the restricted model with that of the full model using the likelihood ratio test.
Longitudinal genetic analyses
The change in genetic and environmental influences on symptoms of AAD, and specifically whether genetic influences could be best described by the developmentally dynamic or by the developmentally stable hypothesis, was explored by modeling the data in a longitudinal ACE model. The longitudinal model, which examines the cross-age correlations as well as the within-age correlations, has more power to discriminate between additive genetic and shared environmental factors than the cross-sectional models, which only examine the within-age correlations. In the longitudinal model, one threshold (six in total) and one deviation from the threshold for females (six in total) were estimated in each age group.
In the saturated (full ACE) model the variance–covariance structure was first explored with a Cholesky decomposition (6 A factors, 6 C factors, 6 E factors) (model 1). In model 2, the sex effects on the thresholds were constrained to be equal over the six age groups. Simplex models were specified to test whether the genetic and environmental influences on AAD symptoms showed qualitative or quantitative change. The final model was built by testing a series of restricted submodels. The unique environmental structure was first examined, followed by the development of shared environmental influences and finally by the structure of the genetic influences. All submodels were compared to the preceding model, with the likelihood ratio test.
First, in submodel 3a a unique environmental simplex structure was specified (Guttman 1955; Boomsma and Molenaar 1987) that allowed for both stable and dynamic influences. This model contained six latent unique environmental variance components, each with one observed measurement as its (only) indicator. The unique environmental variance underlying the observed measurement at a particular age group was determined by the transmission terms (ß) that describe the amplification or attenuation of the unique environmental variance that was already present in the previous age group (E i−1 ), plus the innovation terms (ζ) that allow for new unique environmental variance ‘coming on line’: E i = ße 2 i × E i−1 + ζe 2 i (Neale and Cardon 1992). Since 15–17 was the first age group, no unique environmental variance could be transmitted from an earlier age group. Therefore, the unique environmental variance for this age group reduced to ζe 21 . For the CAGE at age 18–20, the unique environmental variance equaled ße 21 × ζe 21 + ζe 22 and for age 21–23 ße 22 × (ße 21 × ζe 21 + ζe 22 ) + ζe 23 . The unique environmental covariance over age is modeled to run via the transmission terms only and can therefore be separated from variance due to measurement error that is modeled to influence the variances but not the covariances. For model identification purposes, a constraint has to be applied to the measurement error variance at the outer categories (Neale and Cardon 1992). In this study, variance due to measurement error was constrained to be the same for all age groups (submodel 3a). Whether unique environmental influences were purely age-specific was tested by fixing the transmission terms at zero in submodel 3b.
Secondly, a simplex structure was specified for shared environmental influences allowing for both stable and dynamic influences (submodel 4a). Next, in submodel 4b the shared environmental innovation terms were dropped from the model to test whether shared environmental influences could be described by one factor.
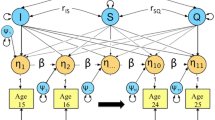
Thirdly, the genetic structure was investigated. In submodel 5a a simplex structure was specified for genetic factors including transmission and innovation terms that allowed for both stable and dynamic genetic influences. Subsequently, in submodel 5b, the innovation terms were dropped from the model allowing for stable genetic influences only. The simplex structure including transmission and innovation terms is shown in Fig. 1a and the model without the innovation terms is shown in Fig. 1b.

Representation of fitted models for analyzing six age groups (in squares) (shown for one twin only). a Simplex structure with six latent variance components (A 1 –A 6 ), five transmission terms (ß 1 –ß 5) and six innovation terms (ζ a 1 –ζ a 6 ). b Simplex structure without innovation
Results
Measurement invariance
The configural invariance model, in which the factor loadings and thresholds were estimated freely for each group, had a good fit (χ2 (21) = 19.84, p = 0.531; RMSEA = 0.001). Constraining the factor loadings and thresholds over the groups to be equal was allowed, as indicated by the fit of the strong factorial invariance model (χ2 (36) = 43.66, p = 0.178; RMSEA = 0.017). Restricting the residual variances to be equal over the groups in the strict factorial invariance model was also permitted, based on the RMSEA (χ2 (71) = 119.21, p < 0.001; RMSEA = .031). This means that for different ages and sexes, the same proportion of the variance can be explained by the latent factor. The CAGE, as it is administered in our surveys, is thus measurement invariant over age and sex. Accordingly, answers on the CAGE can be compared from one age and sex to another. With the CAGE being measurement invariant over age and sex, differences in the prevalence are allowed for when these result from a difference at the latent factor level. In an additional model it was examined whether sex differences existed at the latent factor level by testing whether the factor means for males and females could be constrained to be equal. This changed the model fit based on the RMSEA from good into acceptable (Schermelleh-Engel and Moosbrugger 2003) (χ2 (73) = 246.54, p < 0.001; RMSEA = .058), suggesting that sex differences exist at the latent factor level.
Longitudinal and twin correlations
In Table 3 within person-cross age correlations are presented for the dichotomous CAGE scores (0 vs. 1+ yes answers). For intervals up to 12 years, the correlations were of similar magnitude. For intervals larger than 12 years, the correlations started to decrease, but the number of individuals for whom data was available on these large time intervals was relatively small.
The tetrachoric cross-twin correlations are shown in Table 4. The cross twin-within age correlations (presented at the diagonal in italic) showed a different pattern for MZ and DZ twins. MZ correlations were of similar magnitude for the ages 18–29: .40–.50 for MZF and .50–.60 for MZM. At age 15–17 the correlations were somewhat higher (r MZM = .73; r MZF = .54) and for MZM at age 30–32 somewhat lower (.35). DZ correlations started high at age 15–17 (r DZM = .68; r DZF = .56; r DOS = .48), but were much lower at later ages. This was seen most clearly for DZM, but was also true for DZF and DOS twins. This pattern suggests the influence of shared environmental factors at younger ages and the increase of importance of genetic influences over time.
The cross twin-cross age correlations (shown off-diagonal in Table 4) were lower than the within person-cross age correlations. Overall, cross twin-cross age correlations also decreased over age, although the pattern was less consistent than that seen for the within person correlations.
Table 5 provides the results for testing sex differences in the correlations cross-sectionally. For all age groups, male and female MZ correlations and male DZ, female DZ and DZ opposite sex correlations could be constrained to be equal. Therefore, Table 5 shows the pooled correlations over sex, together with thresholds estimates and deviations from the threshold for females. As described in the section on measurement invariance, there was some indication for true sex differences in CAGE scores (i.e., sex differences that were not due to differences in measurement across sex). To account for these differences, sex effects on the thresholds were tested. Dropping the sex effect on the threshold led to a significantly worse model fit in all age groups (15–17: χ2 (1) = 15.86, p < 0.001; 18–20: χ2 (1) = 109.33, p < 0.001; 21–23: χ2 (1) = 153.15, p < 0.001; 24–26: χ2 (1) = 157.37, p < 0.001; 27–29: χ2 (1) = 67.86, p = 0.001; and 30–32: χ2 (1) = 59.65, p < 0.001). This indicates that the prevalence of giving one or more positive answers differed for men and women. Men more often gave a positive answer on one or more CAGE items than women.
Table 5 also presents the twin correlations and threshold estimates for the trichotomous CAGE (0 vs. 1 vs. 2+ yes answers). The estimates for the dichotomous (0 vs. 1+ yes answers) and trichotomous CAGE (0 vs. 1 vs. 2+ yes answers) were highly similar. Thus, dichotomizing the CAGE using one positive item as cut-off did not bias the observed correlations, nor the thresholds estimates or deviation from the threshold for females as compared to analyzing the CAGE using three response categories. Therefore, further results are presented for the dichotomous CAGE only.
Cross-sectional genetic analyses
Table 6 shows the ACE or ADE variance components estimates and model fitting results for the cross-sectional genetic analyses. Since the MZ correlation was smaller than twice the DZ correlation for the age groups 15–17, 18–20 and 30–32, the ACE model was fitted for these age groups. For the age groups 21–23, 24–26, and 27–29, the MZ correlation was more than twice the DZ correlation and thus the ADE model was fitted. Comparison of the full models showed an increase of unique environmental factors over time, explaining 38% of the variance in AAD symptoms at age 15–17 and 59% at age 30–32. Shared environmental influences decreased over time. While almost half of the variance (45%) was explained by shared environmental influences at age 15–17, and 19% of the variance at age 18–20, no shared environmental effects were present for the ages 21–29. At age 30–32, shared environmental influences accounted for 12% of the variance again. Genetic influences showed an increase in importance over age. At age 15–20, 18% of the variance in AAD symptoms was explained by genetic influences. At age 18–20, this increased to 28% and for the ages 21–29, 50% of the variance could be explained by genetic factors. At age 30–32 genetic influences were less important, explaining 28% of the variance.
For all ages the reduced AE model did not fit significantly worse than the ADE model (model fit statistics shown in Table 6). At the ages 15–17, 18–20 and 30–32, also a reduced CE model fitted the data well, indicating that familial factors were clearly important for this age. Since the AE model described the data well for all age groups, but the CE model was also possible at ages 18–20 and 30–32, the ACE model was selected for the longitudinal analyses.
Longitudinal genetic analyses
Table 7 presents the model fit results for the longitudinal genetic analyses. Constraining the sex effects (differing thresholds for males and females) to be equal over the six age groups deteriorated the model fit significantly as compared to the saturated model (model 2: χ2(5) = 17.69, p = 0.003). Therefore, in subsequent models separate sex effects for the female threshold deviation from the threshold for males were retained for the different age groups.
The unique environmental simplex model with innovation and transmission terms fitted the data well (model 3a: χ2 (9) = 7.78, p = 0.557). Fixing the transmission terms at zero and thereby imposing the matrix with unique environmental factors to be diagonal worsened the model fit (model 3b: χ2 (5) = 26.66, p < .001). This indicates that unique environmental covariance is present over time and shows that both stable and dynamic unique environmental influences impact the CAGE.
Subsequent modeling of the shared environmental influences showed that the simplex model including both transmission and innovation terms (model 4a; χ2 (10) = 3.45, p = 0.969) fitted the data well. Dropping the shared environmental innovation terms from the model was allowed (model 4b; χ2 (5) = 0.17, p = 0.999) indicating that shared environmental innovation was not present and that one shared environmental factor influenced the CAGE.
Finally, fitting a genetic simplex model with transmission and innovation terms did not worsen the model fit (model 5a: χ2 (10) = 11.62, p = 0.311). The final model without genetic innovation terms did not fit significantly worse than the model with genetic innovation (model 5b: χ2 (5) = 8.81, p = 0.147). This final model showed that one stable genetic factor was influencing the CAGE over age.
The parts of the (co)variance that can be explained by additive genetic, shared environmental and unique environmental factors as estimated in the final model are presented in Table 8. Results suggested that 25% of the variance at a particular age could be accounted for by measurement error. In Fig. 2 the associated parameter estimates of this model are shown.

Parameter estimates in final model including measurement error: ACE simplex model without innovation for A and C with innovation for E (shown for one twin only)
When taking variance due to measurement error into account (letting A + C+E sum up to 100%), it can be seen that shared environmental influences showed rapid attenuation over time. While explaining 57% of the variance at age 15–17, (almost) no environmental effects were present anymore for the ages 21–32. Genetic factors showed quantitative changes in its influences over time, increasing from 28% at age 15–17 to 58% at age 21–23. Genetic influences remained high at later ages, accounting for still 52% of the variance at age 30–32. The influence of unique environmental influences was small at age 15–17 (15%), but amplified during the next years. At age 30–32 unique environmental influences were as important as additive genetic influences in explaining differences in the CAGE. Interestingly, phenotypic stability and change in the CAGE over time could be largely attributed to additive genetic factors, although at later ages unique environmental factors also contributed to the stability and change.
For unique environmental influences a simplex structure with innovation terms was specified, allowing for stable and dynamic influences. At younger ages the largest part of unique environmental influences was due to unique environmental innovation. According to the final model, at age 18–20, 98% of the unique environmental variance was due to innovation (.582/(.342 × .272 + .582) × 100%). At age 21–23, this was 83% (.502/[(.342 × .272 + .582) × .382 + .502] × 100%) and at age 24–26, 65% (.472/([(.342 × .272 + .582) × .382 + .502] × .632 + .472) × 100%). At later ages, the largest part of unique environmental influences consisted of those experienced earlier and transmitted to a later age. For example, at age 27–29, only 16% of unique environmental variance was due to innovation (.232/[([(.342 × .272 + .582) × .382 + .502] × .632 + .472) × .912 + .232] × 100%) and 84% to unique environmental factors experienced earlier in life. For age 27–29, 72% of unique environmental influences consisted of those transmitted from earlier ages and 28% was due to innovation (.322/([([(.342 × .272 + .582) × .382 + .502] × .632 + .472) × .912 + .232] × .892 + .322) × 100%).
Discussion
Our main goal was to study the temporal pattern of genetic factors for AAD symptoms during the critical developmental period from adolescence into early adulthood. Studying age-related changes was allowed, since the CAGE assessed the same phenotype across age 15–32 and across men and women, as indicated by the analyses on measurement invariance. In particular, we wanted to discriminate between the developmentally stable and developmentally dynamic hypotheses that predict quite different patterns for cross age genetic effects on symptoms of AAD. Genetic innovation was not detected, which provides evidence in favor of the developmentally stable hypothesis. That is, genetic risk factors for symptoms of AAD appear to be temporally stable across the key developmental period from mid-adolescence into early adulthood. The same genetic risk factors are important at younger and older ages. This is in line with Sartor et al. (2008) who found that one common genetic factor could explain the rate of progression from age at alcohol initiation to age at occurrence of the first symptom of AAD and age at onset of an alcohol dependence diagnosis. Although in our study no qualitative changes in genetic factors were found, quantitative changes were present. Genetic influences amplified from age 15–17 to age 21–23 and then remained high.
Genetic factors can impact on risk for AAD symptoms at a wide range of levels and are likely to share genetic influences on alcohol consumption measures, partly (Whitfield et al. 2004) or (almost) entirely (Grant et al. 2009; Kendler et al. 2010). Genetic influences include those on alcohol metabolism by the ADH and ALDH gene cluster (Edenberg et al. 2006; Luo et al. 2006; Kuo et al. 2008; Macgregor et al. 2009; van Beek et al. 2010), the level of response to alcohol (Schuckit et al. 2004) and neurotransmitters related to the reward system that interact directly and indirectly with ethanol (e.g., dopamine, and GABA) (Enoch 2008; Smith et al. 2008; Munafo et al. 2007). Genetic influences on AAD also act via general predispositions to abuse licit and illicit psychoactive drugs (Kendler et al. 2007; Agrawal et al. 2008), predispositions toward externalizing behaviors in general (Cerda et al. 2010) and the tendency to self-select into environments where alcohol is widely available and its misuse socially condoned (Kendler et al. 2007; Gillespie et al. 2007). In addition, genetic factors for AAD can have their influence by acting on co-morbid psychiatric symptomatology, such as depression or anxiety disorders (Cerda et al. 2010; Saraceno et al. 2009).
This project had the additional goal of clarifying the change in environmental influences on symptoms of AAD between the ages 15 and 32. A single shared environmental factor had a large influence on AAD symptoms at age 15–17, but quickly attenuated over time. Consistent with Dick et al. (2009) shared environmental influences on symptoms of AAD are important in adolescence, but less (during age 18–23) or not (from 24–26 onwards) at later ages. Attenuation of shared environmental influences in this period of life is observed for other phenotypes as well, such as for IQ (Segal and Johnson 2009), social attitudes and nicotine initiation (Bergen et al. 2007) and makes sense from a developmentally point of view in which individuals of this age tend to leave the parental home and sometimes also the neighborhood. The shared environmental effects could be a reflection of parenting practices (Latendresse et al. 2010), drinking family members (Poelen et al. 2007; Mcgue et al. 1996), or religious upbringing (Kendler and Myers 2009). The home environment that parents provide for their children might also in part reflect the genes the parents transmit to their children. If parents transmit both genes and environment—referred to as genetic and cultural transmission—this induces a passive correlation between G and C (e.g., Rijsdijk and Sham 2002). Passive G–C correlation might for instance occur if one of the parents suffers from AAD which also creates an environment in which the availability of alcohol at home is more likely. In addition to familial influences originating at home, the shared environmental effects in twins might also reflect the influence of peers’ alcohol use when twins share the same peer group (Fowler et al. 2007).
The cross-sectional analyses suggested that shared environmental influences were also present at age 30–32 in close correspondence with Kendler et al. (2008c) who found shared environmental effects for alcohol consumption for roughly the same age. For people of this age, who commonly start a family life in this period of life (CBS 2010), a decrease in alcohol consumption is not surprising. In particular in women (62% of our sample), alcohol consumption will change when being pregnant or breastfeeding. Therefore, at least for women, the shared environmental effects at age 30–32 might reflect a decrease in alcohol consumption during pregnancy and breast feeding that is shared within twin pairs, given that age at first birth is partly due to shared environmental influences (Neiss et al. 2002; Rodgers et al. 2008). However, it should be noted that in the longitudinal analyses shared environmental influences were only observed in adolescence and not at age 30–32. It will be interesting to see whether in a few years we can detect evidence for shared environmental factors for people aged 30 and older as we continue our longitudinal data collection and the number of individuals of age 30 and older will increase.
Unique environmental effects increased at age 18–20, the age at which individuals commonly leave the parental home, and remained of similar magnitude at later ages. The variance explained by environmental factors specific to an individual increased from 15% in adolescence to 40–48% during early adulthood. Whereas at younger ages unique environmental influences were largely age-specific, from age 27–29 onwards the largest part of unique environmental influences resulted from those experienced earlier which were then transmitted to a later age. These unique environmental influences can represent any influences that the twins do not have in common, like a specific group of friends, relationships, life events, or working environment.
Our results show that symptoms of AAD are already reported by adolescents. In 15–17 year olds, 15.8% of men and 9% of women reported one or more symptoms. These percentages are not surprising given the high frequency of alcohol consuming adolescents in the Netherlands. Although according to Dutch legislation people have to be aged 16 or older to buy beer and wine and 18 or older for spirits/hard liquor, the rate of ‘success’ for obtaining these substances under the age of 16/18 is estimated at 85%. Three in four adolescents report to have consumed alcohol in the last month at least once and a substantial group even more than 10 times (23% of male, 10% of female alcohol-consuming adolescents) (Trimbos Instituut 2010).
We did not find evidence for genetic innovation although others have. In Malone et al. (2004) genetic innovations were suggested for symptoms of alcohol dependence at age 17, 20 and 24 years. However, they focused on transmission at the phenotypic level, rather than at the level of latent genetic and non-genetic factors. Under this parameterization, a change in heritability is only possible by including a term for genetic innovation. In our analyses, transmission coefficients were specified at the level of latent genetic, common environmental and unique environmental factors. This is a less constrained model that indicated genetic stability without genetic innovation. When analyzing our data with a phenotypic simplex model, genetic innovations were significant (χ2 (5) = 41.30, p < 0.001). This does not reflect new genetic influences however, but the fact that the amounts of genetic, common and unique environmental variance that are transmitted, are not equal to each other. For different phenotypes genetic innovation has been detected, for instance for fears and phobias (Kendler et al. 2008a), anxiety and depression (Kendler et al. 2008b) and externalizing behaviors (Wichers et al. 2011). Our analyses showed that a model without genetic innovation fitted the data as well as a model with genetic innovation. This does not mean that genetic innovation is entirely absent. If we test all genetic innovation parameters separately, parameters at age 21–23 and 24–26 are significant (p = 0.038 and p = 0.009 respectively). Still, our results indicate that the majority of genetic influences are stable over time, and genetic innovation effects are small.
A next step would be searching for susceptibility genes for AAD symptoms. Recently, a number of genome wide association studies have been conducted for alcohol dependence (Bierut et al. 2010; Treutlein et al. 2009; Edenberg et al. 2010; Lind et al. 2010; Kendler et al. 2011). Two closely linked intergenic SNPs at chromosome 2q35 achieved genome-wide significance in the combined analysis of the discovery and follow-up sample in Treutlein et al. (2009). In other studies no SNP met criteria for genome-wide significance (Bierut et al. 2010; Edenberg et al. 2010; Kendler et al. 2011), or genome-wide significant SNPs could not be replicated in a follow-up sample (Lind et al. 2010). Some evidence was found for gene clusters. For instance, in Lind et al. (2010) the top SNPs could be placed in a gene network coding for ion-channels and cell adhesion molecules. Edenberg et al. (2010) found several SNPs on chromosome 11 that were independently marginally associated with alcohol dependence. In regard to the genetic overlap of AAD and alcohol consumption measures (Grant et al. 2009; Kendler et al. 2010), the recent finding of a genome-wide significant association of a SNP at chromosome 7q11.22 with alcohol consumption (Schumann et al. 2011), is also relevant.
The search for susceptibility genes can be aided with a thorough assessment of how genetic influences change across age. When there is no evidence of genetic innovation over time, as for the phenotype under study, a sum score over all available data points might give more power to detect susceptibility loci than analyzing one time point only because sum scores tend to be more reliable than single observations. However, in a scenario in which genetic innovation is present, summing over different observations in time can lead to a considerable drop in power as compared to analyzing one data point only. This was outlined in a simulation study by Minica et al. (2010). With data generated under a one genetic factor model (four time points; heritability of .6; unique environmental effects at .4), the power to detect a genetic risk variant was .96 when a sum score of the four observations was analyzed, and .56 when only one data point was analyzed (α = .01; with a power of .99 for the true model). In the scenario with genetic innovation present and a genetic risk variant that entered the model at the fourth time point (h2 = .6; e2 = .4), the power dropped to .03 when a sum score was analyzed, whereas it stayed at .56 when only observations at the fourth time point were analyzed (α = .01; power true model > .99).
This study includes some potential limitations. First, there is a question of the degree to which the sample is representative. This was examined in two studies on the effect of non-cooperation in the Netherlands Twin Register (Vink et al. 2004; Distel et al. 2007). Vink et al. (2004) found that the proportion of individuals endorsing two or more positive symptoms on the CAGE was greater in families with low vs. high cooperation and in incomplete vs. complete twin pairs. However, the differences were modest in magnitude and not statistically significant after correction for multiple testing. In a later study no difference was detected between high and low cooperative families in the proportion of individuals that scored two positive items or more (Distel et al. 2007). Therefore, the sample is considered to be relatively unbiased with respect to symptoms of AAD. Secondly, the assessment of symptoms of AAD consisted of a four-item self-report questionnaire. The CAGE has been successfully used as a screening instrument for AAD (Dhalla and Kopec 2007; Maisto and Saitz 2003; Aertgeerts et al. 2004) and socially undesirable behaviors such as symptoms of AAD may be better assessed using self-administration methods than interviews (Tipping et al. 2010; Bowling 2005). Lastly, the CAGE inquires about ‘ever’ having symptoms of AAD (Ewing 1984). This would imply that participants cannot answer an item with ‘no’ when they have answered ‘yes’ to that item at an earlier assessment. In total, 8,398 subjects took part in the study and for 3,909 there were at least two observations. In this last group, 5.8% of the replies was inconsistent (determined as the number of inconsistent answers as a function of the total number of replies). That is, a no-reply followed an earlier yes-reply. Giving an inconsistent response was relatively more frequent among males (χ2(1) = 35.30, p < .001), who on average endorse symptoms of AAD more often than women and therefore have a higher chance of giving an inconsistent response, and individuals who entered the study at a younger age (χ2(5) = 133.06, p < .001). Since variance due to measurement error was taken into account in our analyses, the effect of inconsistencies due to measurement error is likely to be minor. It is however likely that certain variability in AAD symptoms has gone undetected by the restriction of the phrasing of the CAGE items to ‘ever’. This might have inflated the stability in symptoms of AAD over time which could have had its effect on our findings of genetic stability.
The absence of genetic innovation might be due to a lack of statistical power resulting from the fact that we used a cut-off score of one or more positive answers such that data from the youngest age group (15–17) could be included also, instead of using a cut-off of two or more yes answers as is usually done. In other populations with a low prevalence of AAD a cut-off score of one or more positive answers was also applied (Buchsbaum et al. 1992). More importantly, the cut-off of ≥1 yes answers did not affect the twin correlations or threshold estimates. To fully address the question whether absence of genetic innovation was due to a lack of power since we used the cut-off of ≥1 positive answers, the analyses were repeated using three response categories (0, 1, 2+ yes answers) and four response categories (0, 1, 2, 3+ yes answers) for the age groups spanning from 18 to 32 years. The youngest age group was not included, since in this group the frequency of two or more yes answers was too low. Including more response categories did not change the results. Dropping the terms for genetic innovation did not worsen the model fit significantly for the CAGE as trichotomy (χ2 (4) = 0.530; p = 0.971), nor when the CAGE was analyzed with four response categories (χ2(4) = 0.681, p = .954). This renders lack of power as an explanation for the absence of genetic innovation, unlikely.
In conclusion, genetic influences on symptoms of AAD in adolescence and young adulthood are best described by the developmentally stable hypothesis. Symptoms of AAD are influenced by stable genetic risk factors and environmental influences that are largely age-specific.
References
Aertgeerts B, Buntinx F, Kester A (2004) The value of the CAGE in screening for alcohol abuse and alcohol dependence in general clinical populations: a diagnostic meta-analysis. J Clin Epidemiol 57:30–39
Agrawal A, Hinrichs AL, Dunn G, Bertelsen S, Dick DM, Saccone SF et al (2008) Linkage scan for quantitative traits identifies new regions of interest for substance dependence in the Collaborative Study on the Genetics of Alcoholism (COGA) sample. Drug Alcohol Depend 93:12–20
Bartels M, van Beijsterveldtl CEM, Derks EM, Stroet TM, Polderman TJC, Hudziak JJ et al (2007) Young Netherlands Twin Register (Y-NTR): a longitudinal multiple informant study of problem behavior. Twin Res Hum Genet 10:3–11
Bergen SE, Gardner CO, Kendler KS (2007) Age-related changes in heritability of behavioral phenotypes over adolescence and young adulthood: a meta-analysis. Twin Res Hum Genet 10:423–433
Bierut LJ, Agrawal A, Bucholz KK, Doheny KF, Laurie C, Pugh E et al (2010) A genome-wide association study of alcohol dependence. Proc Natl Acad Sci USA 107:5082–5087
Boomsma DI, Molenaar PCM (1987) The genetic-analysis of repeated measures.1. Simplex models. Behav Genet 17:111–123
Boomsma DI, Vink JM, van Beijsterveldt TCEM, de Geus EJC, Beem AL, Mulder EJCM et al (2002) Netherlands Twin Register: a focus on longitudinal research. Twin Res 5:401–406
Boomsma DI, de Geus EJC, Vink JM, Stubbe JH, Distel MA, Hottenga JJ et al (2006) Netherlands Twin Register: from twins to twin families. Twin Res Hum Genet 9:849–857
Bowling A (2005) Mode of questionnaire administration can have serious effects on data quality. J Public Health 27:281–291
Buchsbaum DG, Buchanan RG, Welsh J, Centor RM, Schnoll SH (1992) Screening for drinking disorders in the elderly using the cage questionnaire. J Am Geriatr Soc 40:662–665
CBS (Statistics Netherlands) (2010) http://statline.cbs.nl/StatWeb/publication/?DM=SLNL&PA=37620&D1=4&D2=0&D3=102-104&D4=0,5,10,(l-2)-l&VW=T. Accessed 1 July 2011
Cerda M, Sagdeo A, Johnson J, Galea S (2010) Genetic and environmental influences on psychiatric comorbidity: a systematic review. J Affect Disord 126:14–38
Cloninger CR, Bohman M, Sigvardson S (1981) Inheritance of alcohol-abuse—cross-fostering analysis of adopted men. Arch Gen Psychiatry 38:861–868
Dhalla S, Kopec JA (2007) The CAGE questionnaire for alcohol misuse: a review of reliability and validity studies. Clin Investig Med 30:33–41
Dick DM, Prescott CA, Mcgue M (2009) The genetics of substance use and substance use disorders. In: Kim Y-K (ed) Handbook of behavior genetics, 1st edn. Springer, New York, pp 433–453
Distel MA, Ligthart L, Willemsen G, Nyholt DR, Trull TJ, Boomsma DI (2007) Personality, health and lifestyle in a questionnaire family study: a comparison between highly cooperative and less cooperative families. Twin Res Hum Genet 10:348–353
Edenberg HJ, Xuei XL, Chen HJ, Tian HJ, Wetherill LF, Dick DM et al (2006) Association of alcohol dehydrogenase genes with alcohol dependence: a comprehensive analysis. Hum Mol Genet 15:1539–1549
Edenberg HJ, Koller DL, Xuei X, Wetherill L, McClintick JN, Almasy L et al (2010) Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol Clin Exp Res 34:840–852
Enoch MA (2008) The role of GABA(A) receptors in the development of alcoholism. Pharmacol Biochem Behav 90:95–104
Ewing JA (1984) Detecting alcoholism—the cage questionnaire. J Am Med Assoc 252:1905–1907
Fowler T, Shelton K, Lifford K, Rice F, McBride A, Nikolov I et al (2007) Genetic and environmental influences on the relationship between peer alcohol use and own alcohol use in adolescents. Addiction 102:894–903
Geels LM, Bartels M, van Beijsterveldt CEM, Willemsen G, Van der Aa N, Vink JM et al (2011) Trends in adolescent alcohol use: effects of age, sex and cohort on prevalence and heritability (under revision)
Gillespie NA, Kendler KS, Prescott CA, Aggen SH, Gardner CO, Jacobson K et al (2007) Longitudinal modeling of genetic and environmental influences on self-reported availability of psychoactive substances: alcohol, cigarettes, marijuana, cocaine and stimulants. Psychol Med 37:947–959
Goodwin DW, Schulsin F, Hermanse L, Guze SB, Winokur G (1973) Alcohol problems in adoptees raised apart from alcoholic biological parents. Arch Gen Psychiatry 28:238–243
Grant JD, Agrawal A, Bucholz KK, Madden PAF, Pergadia ML, Nelson EC et al (2009) Alcohol consumption indices of genetic risk for alcohol dependence. Biol Psychiatry 66:795–800
Guttman L (1955) A new approach to factor analysis: the radex. In: Lazarsfeld PF (ed) Mathematical thinking in the social sciences, 2nd edn. The Free Press, Glencoe, pp 258–348
Harford TC, Grant BF, Yi HY, Chen CM (2005) Patterns of DSM-IV alcohol abuse and dependence criteria among adolescents and adults: results from the 2001 National Household Survey on Drug Abuse. Alcohol Clin Exp Res 29:810–828
Heath AC, Bucholz KK, Madden PAF, Dinwiddie SH, Slutske WS, Bierut LJ et al (1997) Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychol Med 27:1381–1396
Kendler KS, Myers J (2009) A developmental twin study of church attendance and alcohol and nicotine consumption: a model for analyzing the changing impact of genes and environment. Am J Psychiatry 166:1150–1155
Kendler KS, Heath AC, Neale MC, Kessler RC, Eaves LJ (1992) A population-based twin study of alcoholism in women. J Am Med Assoc 268:1877–1882
Kendler KS, Prescott CA, Neale MC, Pedersen NL (1997) Temperance board registration for alcohol abuse in a national sample of Swedish male twins, born 1902 to 1949. Arch Gen Psychiatry 54:178–184
Kendler KS, Myers J, Prescott CA (2007) Specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine, and nicotine dependence. Arch Gen Psychiatry 64:1313–1320
Kendler KS, Gardner CO, Annas P, Neale MC, Eaves LJ, Lichtenstein P (2008a) A longitudinal twin study of fears from middle childhood to early adulthood—evidence for a developmentally dynamic genome. Arch Gen Psychiatry 65:421–429
Kendler KS, Gardner CO, Lichtenstein P (2008b) A developmental twin study of symptoms of anxiety and depression: evidence for genetic innovation and attenuation. Psychol Med 38:1567–1575
Kendler KS, Schmitt E, Aggen SH, Prescott CA (2008c) Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry 65:674–682
Kendler KS, Myers J, Dick D, Prescott CA (2010) The relationship between genetic influences on alcohol dependence and on patterns of alcohol consumption. Alcohol Clin Exp Res 34:1058–1065
Kendler KS, Kalsi G, Holmans P, Sanders A, Aggen SH, Dick D et al (2011) Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol Clin Exp Res 35:963–975
Knopik VS, Heath AC, Madden PAF, Bucholz KK, Slutske WS, Nelson EC et al (2004) Genetic effects on alcohol dependence risk: re-evaluating the importance of psychiatric and other heritable risk factors. Psychol Med 34:1519–1530
Koopmans JR, Boomsma DI (1996) Familial resemblances in alcohol use: genetic or cultural transmission? J Stud Alcohol 57:19–28
Koppes LLJ, Kemper HCG, Post GB, Snel J, Twisk JWR (2000) Development and stability of alcohol consumption from adolescence into adulthood: the Amsterdam growth and health longitudinal study. Eur Addict Res 6:183–188
Kuo PH, Kalsi G, Prescott CA, Hodgkinson CA, Goldman D, van den Oord EJ et al (2008) Association of ADH and ALDH genes with alcohol dependence in the Irish affected sib pair study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res 32:785–795
Latendresse SJ, Rose RJ, Viken RJ, Pulkkinen L, Kaprio J, Dick DM (2010) Examining the etiology of associations between perceived parenting and adolescents’ alcohol use: common genetic and/or environmental liabilities? J Stud Alcohol Drugs 71:313–325
Lind PA, Macgregor S, Vink JM, Pergadia ML, Hansell NK, de Moor MHM et al (2010) A genomewide association study of nicotine and alcohol dependence in Australian and Dutch populations. Twin Res Hum Genet 13:10–29
Liu IC, Blacker DL, Xu RH, Fitzmaurice G, Lyons MJ, Tsuang MT (2004) Genetic and environmental contributions to the development of alcohol dependence in male twins. Arch Gen Psychiatry 61:897–903
Luo XG, Kranzler HR, Zuo LJ, Lappalainen J, Yang BZ, Gelernter J (2006) ADH4 gene variation is associated with alcohol dependence and drug dependence in European Americans: results from HWD tests and case-control association studies. Neuropsychopharmacology 31:1085–1095
Macgregor S, Lind PA, Bucholz KK, Hansell NK, Madden PAF, Richter MM et al (2009) Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: an integrated analysis. Hum Mol Genet 18:580–593
Maisto SA, Saitz R (2003) Alcohol use disorders: screening and diagnosis. Am J Addict 12:S12–S25
Malone SM, Taylor J, Marmorstein NR, Mcgue M, Iacono WG (2004) Genetic and environmental influences on antisocial behavior and alcohol dependence from adolescence to early adulthood. Dev Psychopathol 16:943–966
Mcgue M, Sharma A, Benson P (1996) Parent and sibling influences on adolescent alcohol use and misuse: evidence from a US adoption cohort. J Stud Alcohol 57:8–18
Midanik LT, Clark WB (1994) The demographic distribution of US drinking patterns in 1990—description and trends from 1984. Am J Public Health 84:1218–1222
Minica CC, Boomsma DI, van der Sluis S, Dolan CV (2010) Genetic association in multivariate phenotypic data: power in five models. Twin Res Hum Genet 13:525–543
Moore AA, Gould R, Reuben DB, Greendale GA, Carter K, Zhou KF et al (2005) Longitudinal patterns and predictors of alcohol consumption in the United States. Am J Public Health 95:458–464
Munafo MR, Matheson IJ, Flint J (2007) Association of the DRD2 gene Taq1A polymorphism and alcoholism: a meta-analysis of case-control studies and evidence of publication bias. Mol Psychiatry 12:454–461
Muthén LK, Muthén BO (2007) Mplus user’s guide, 5th edn. Muthén & Muthén, Los Angeles
Neale MC, Cardon LR (1992) Methodology for genetic studies of twins and families. Kluwer, Dordrecht, pp 289–304
Neale, MC, Boker, SM, Xie, G, & Maes, HH (2006) Mx: statistical modeling, 7th edn. Department of Psychiatry, VCU, Richmond
Neiss M, Rowe DC, Rodgers JL (2002) Does education mediate the relationship between IQ and age of first birth? A behavioural genetic analysis. J Biosoc Sci 34:259–275
Pickens RW, Svikis DS, Mcgue M, Lykken DT, Heston LL, Clayton PJ (1991) Heterogeneity in the inheritance of alcoholism—a study of male and female twins. Arch Gen Psychiatry 48:19–28
Poelen EA, Scholte RH, Engels RC, Boomsma DI, Willemsen G (2005) Prevalence and trends of alcohol use and misuse among adolescents and young adults in the Netherlands from 1993 to 2000. Drug Alcohol Depend 79:413–421
Poelen EAP, Scholte RHJ, Willemsen G, Boomsma DI, Engels RCME (2007) Drinking by parents, siblings, and friends as predictors of regular alcohol use in adolescents and young adults: a longitudinal twin-family study. Alcohol Alcohol 42:362–369
Prescott CA, Kendler KS (1999) Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. Am J Psychiatry 156:34–40
R Development Core Team (2010) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rebollo I, de Moor MHM, Dolan CV, Boomsma DI (2006) Phenotypic factor analysis of family data: correction of the bias due to dependency. Twin Res Hum Genet 9:367–376
Reed T, Page WF, Viken RJ, Christian JC (1996) Genetic predisposition to organ-specific endpoints of alcoholism. Alcohol Clin Exp Res 20:1528–1533
Rijsdijk FV, Sham PC (2002) Analytic approaches to twin data using structural equation models. Brief Bioinform 3:119–133
Rodgers JL, Kohler HP, Mcgue M, Behrman JR, Petersen I, Bingley P et al (2008) Education and cognitive ability as direct, mediating, or spurious influences on female age at first birth: behavior genetic models fit to danish twin data. Am J Sociol 114:S202–S232
Rose RJ, Dick DM, Viken RJ, Kaprio J (2001) Gene-environment interaction in patterns of adolescent drinking: regional residency moderates longitudinal influences on alcohol use. Alcohol Clin Exp Res 25:637–643
Saraceno L, Munafo M, Heron J, Craddock N, van den Bree MBM (2009) Genetic and non-genetic influences on the development of co-occurring alcohol problem use and internalizing symptomatology in adolescence: a review. Addiction 104:1100–1121
Sartor CE, Agrawal A, Lynskey MT, Bucholz KK, Heath AC (2008) Genetic and environmental influences on the rate of progression to alcohol dependence in young women. Alcohol Clin Exp Res 32:632–638
Sartor CE, Grant JD, Bucholz KK, Madden PAF, Heath AC, Agrawal A et al (2010) Common genetic contributions to alcohol and cannabis use and dependence symptomatology. Alcohol Clin Exp Res 34:545–554
Schermelleh-Engel K, Moosbrugger H (2003) Evaluating the fit of structural equation models: tests of significance and descriptive goodness-of-fit measures. Methods Psychol Res Online 8:23–74
Schuckit MA, Daeppen JB, Tipp JE, Hesselbrock M, Bucholz KK (1998) The clinical course of alcohol-related problems in alcohol dependent and nonalcohol dependent drinking women and men. J Stud Alcohol 59:581–590
Schuckit MA, Smith TL, Kalmijn J (2004) The search for genes contributing to the low level of response to alcohol: patterns of findings across studies. Alcohol Clin Exp Res 28:1449–1458
Schumann G, Coin LJ, Lourdusamy A, Charoen P, Berger KH, Stacey D et al (2011) Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci USA 108:7119–7124
Segal NL, Johnson W (2009) Twin studies of general mental ability. In: Kim Y-K (ed) Handbook of behavior genetics, 1st edn. Springer, New York, pp 81–100
Slof-Op ‘t Landt MCT, Dolan CV, Rebollo-Mesa I, Bartels M, van Furth EF, van Beijsterveldt CEM et al (2009) Sex differences in sum scores may be hard to interpret: the importance of measurement invariance. Assessment 16:415–423
Smith L, Watson M, Gates S, Ball D, Foxcroft D (2008) Meta-analysis of the association of the Taq1A polymorphism with the risk of alcohol dependency: a HuGE gene-disease association review. Am J Epidemiol 167:125–138
Tipping S, Hope S, Pickering K, Erens B, Roth MA, Mindell JS (2010) The effect of mode and context on survey results: analysis of data from the health survey for England 2006 and the boost survey for London. BMC Med Res Methodol 10:84
Treutlein J, Cichon S, Ridinger M, Wodarz N, Soyka M, Zill P et al (2009) Genome-wide association study of alcohol dependence. Arch Gen Psychiatry 66:773–784
Trimbos Instituut (Netherlands Institute of Mental Health and Addiction) (2010) National Drug Monitor 2009. http://www.rijksoverheid.nl/documenten-en-publicaties/rapporten/2010/05/27/nationale-drugmonitor-2009.html. Accessed 28 Apr 2011
True WR, Xian H, Scherrer JF, Madden PAF, Bucholz KK, Heath AC et al (1999) Common genetic vulnerability for nicotine and alcohol dependence in men. Arch Gen Psychiatry 56:655–661
van Beek JHDA, Willemsen G, de Moor MHM, Hottenga JJ, Boomsma DI (2010) Associations between ADH gene variants and alcohol phenotypes in Dutch adults. Twin Res Hum Genet 13:30–42
Viken RJ, Kaprio J, Koskenvuo M, Rose RJ (1999) Longitudinal analyses of the determinants of drinking and of drinking to intoxication in adolescent twins. Behav Genet 29:455–461
Vink JM, Willemsen G, Stubbe JH, Middeldorp CM, Ligthart RSL, Baas KD et al (2004) Estimating non-response bias in family studies: application to mental health and lifestyle. Eur J Epidemiol 19:623–630
Whitfield JB, Zhu G, Madden PA, Neale MC, Heath AC, Martin NG (2004) The genetics of alcohol intake and of alcohol dependence. Alcohol Clin Exp Res 28:1153–1160
Wichers M, Gardner C, Maes HH, Lichtenstein P, Larsson H, Kendler KS (2011) Genetic innovation and stability in externalising problem behavior across development: a multi-informant twin study (under revision)
Willemsen G, Posthuma D, Boomsma DI (2005) Environmental factors determine where the Dutch live: results from the Netherlands Twin Register. Twin Res Hum Genet 8:312–317
Acknowledgments
This work was supported by grants from the Netherlands Organization for Scientific Research (ZonMW Addiction 31160008; ZonMW 940-37-024; NWO/SPI 56-464-14192; NWO-400-05-717; NWO-MW 904-61-19; and NWO-MagW 480-04-004), the European Research Council (Genetics of Mental Illness: ERC-230374) and the NIH (AA-011408 and DA-011287). We want to thank dr. C.V. Dolan for his statistical support and all the twin families for their participation in the NTR research.
Conflicts of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Chandra Reynolds.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Beek, J.H.D.A., Kendler, K.S., de Moor, M.H.M. et al. Stable Genetic Effects on Symptoms of Alcohol Abuse and Dependence from Adolescence into Early Adulthood. Behav Genet 42, 40–56 (2012). https://doi.org/10.1007/s10519-011-9488-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-011-9488-8