
Abstract
The purpose of this investigation was to evaluate the diversity of bacteria in diabetic foot osteomyelitis using a 16S rRNA sequencing approach and to compare the results with conventional culture techniques. In this prospective observational study, we obtained 34 bone samples from patients admitted to our hospital with a moderate–severe diabetic foot infection. We analysed the distribution of the 16S rRNA gene sequences in the bone samples, using an Illumina MiSeq Personal Sequencer. We compared the genera that were detected with the cultured pathogens in the bone samples with conventional techniques. In the 23 samples that had positive results with both techniques, Staphylococcus, Corynebacterium, Streptococcus and Propionibacterium spp. were detected in 20, 18, 13 and 11 samples, respectively. Significantly more anaerobes were detected with 16S rRNA sequencing compared to conventional techniques (86.9 % vs. 23.1 %, p = 0.001) and more Gram-positive bacilli were present (78.3 % vs. 3.8 %, p < 0.001). Staphylococcus spp. were identified in all of the sequenced bone samples that were negative with conventional techniques. Mixed genera were present in 83.3 % (5 of 6) of the negative samples. Anaerobic and fastidious organisms may play a more significant role in osteomyelitis than previously reported. Further studies with larger populations are needed in order to fully understand the clinical importance of the microbial diversity of diabetic foot osteomyelitis.
Similar content being viewed by others
Introduction
Diabetic foot osteomyelitis (DFO) develops in approximately 44–68 % of patients with diabetes mellitus admitted to the hospital with a diabetic foot infection (DFI) [1] and is the leading cause of amputation among such patients [2]. The microbiologic spectrum of DFO seems to be similar to deep diabetic foot soft tissue infections [3] and primarily consists of Gram-positive bacteria, especially Staphylococcus aureus and beta haemolytic streptococci [4, 5]. Anaerobic pathogens are generally uncommon, with some studies reporting that only 3–14 % of infections involve anaerobes [6]. More recent studies indicate that 46–85 % of DFO are monomicrobial [7, 8]. However, conventional culture techniques focus on organisms easily cultured using traditional microbiological evaluations and are limited by the time required for organisms to grow [9]. The phenomenon that only a small percentage of microorganisms grow on agar plates has been known as the ‘great plate count anomaly’ since the early 20th century [10]. Little is known about the diversity of bacteria in DFO and the contribution of anaerobic and fastidious organisms to these infections [11]. This study aimed to better characterise the bacterial ecology of DFO using a modern 16S ribosomal ribonucleic acid (rRNA) gene sequencing approach.
Materials and methods
Patient population
We consecutively obtained 34 bone samples from patients admitted to our hospital with moderate–severe DFI according to the diabetic foot infection classification of the Infectious Diseases Society of America (IDSA) [12]. We included patients who were 21 years or older and had high suspicion of DFO based on their IDSA classification. Exclusion criteria included other infectious diseases, active, previously diagnosed DFO in the study foot, immunosuppressive therapy, organ and/or haematological malignancies, and end-stage renal disease requiring dialysis. We performed a percutaneous biopsy using a 16 gauge Jamshidi needle introduced at least 2 cm from the ulcer site [6] (n = 7) or we obtained intraoperative bone samples from the patients that required surgical debridement or amputation (n = 27). We sent the obtained bone samples to the laboratory for conventional culturing and histopathological tests. We used our hospital microbiology laboratory’s established protocol for anaerobic sampling and transport. The bone specimens were placed in sterile cups without any transport medium and processed within 1 h of collection. Laboratory technicians were kept unaware of the clinical data. We stored a part of the obtained samples promptly at −80 °C until the end of the study. Informed consent was obtained from all individual participants included in the study.
16S rRNA gene sequencing
After thawing, we recovered portions of the bone samples using sterile forceps. We extracted genomic DNA using the Roche High Pure PCR Template Preparation Kit (Roche Life Sciences, Indianapolis, IN, USA) with a modified lysis step. We lysed our samples by combining 25 mg of sample, 200 μL each of lysis buffer and binding buffer (kit buffers 1 and 2), 500 μL of zirconium oxide beads and a single 5-mm steel bead in a 200-μL screw-cap tube. We shook the tubes using a TissueLyser II (Qiagen, Inc., Valencia, CA, USA) for 5 min at 30 Hz. We continued the extraction using the manufacturer’s instructions. We used the Illumina MiSeq Personal Sequencer (Illumina, Inc., San Diego, CA, USA) in collaboration with PathoGenius Laboratories (PathoGenius, Lubbock, TX, USA) to assess the distribution of 16S rRNA gene sequences. We amplified the samples for sequencing using a forward and a reverse fusion primer. The forward primer was constructed with the (5′-3′) Illumina i5 adapter, an 8–10-bp barcode, a primer pad and the 28F primer. The reverse fusion primer was constructed with the (5′-3′) Illumina i7 adapter, an 8–10-bp barcode, a primer pad and the 519R primer. We used the HotStarTaq Plus Master Mix Kit (Qiagen, Inc., Valencia, CA, USA) for polymerase chain reaction (PCR) under the following conditions: 94 °C for 3 min; followed by 30 cycles of 94 °C for 30 s, 60 °C for 40 s and 72 °C for 1 min; and a final elongation step at 72 °C for 5 min.
In preparation for 16S sequencing, we denoised the DNA to remove short sequences, singleton sequences and noisy reads [13]. With the bad reads removed, we performed chimera detection using the de novo method built into UCHIME [14] to aid in the removal of chimeric sequences [15]. We corrected the remaining sequences base by base to help remove noise from within each sequence. We quality checked and demultiplexed the denoised and chimera-checked reads generated during sequencing. We then clustered these sequences into operational taxonomic units (OTUs) using the UPARSE algorithm [16]. We ran the centroid sequence from each cluster against a database of high-quality sequences derived from the National Center for Biotechnology Information (NCBI) database, March 2015. We used an internally developed Python program that assigns taxonomic information to each sequence to analyse the output and write the final analysis files.
Statistical analysis
Analysis was performed using the SAS 9.4 statistical package. The data were presented as number of patients (%). Differences between both culture techniques were measured using the McNemar’s test. p-Values <0.05 were considered statistically significant.
Results
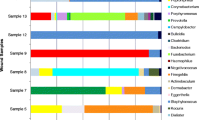
Of the 26 bone samples that grew pathogens with conventional culturing techniques, three did not sequence. The three samples that did not sequence grew Stenotrophomonas maltophilia (n = 1), S. aureus (n = 1) and Enterobacter cloacae (n = 1) with conventional culturing. All three samples were monomicrobial infections. Table 1 presents an overview of all the genera that were sequenced and occurred in at least 21.7 % (5 of 23) of the positive bone samples. The table includes the average contribution of each genus to the total bacterial population in those samples as represented as a percentage. Staphylococcus spp. were the predominant genus identified in the positive bone samples. Sequences of Staphylococcus spp. were detected in 20 of the 23 samples, with an average contribution of 28.6 % to the total bacterial population. The most prevalent populations of Gram-positive cocci identified after that were, in order, Corynebacterium (n = 18), Streptococcus (n = 13) and Propionibacterium spp. (n = 11). Facultative anaerobes included Actinomyces and Helcococcus spp. in 6 and 5 of the samples, respectively. Obligate anaerobes such as Peptoniphilus, Finegoldia, Anaerococcus, Clostridium, Porphyromonas and Prevotella were detected in 17, 15, 12, 9, 7 and 5 of the samples, respectively. Two of the positive samples were low coverage samples (sequence counts of 9 and 534, respectively). Both of these low coverage samples only had sequences that matched with Staphylococcus spp. and grew coagulase-negative staphylococci with conventional techniques.
Table 2 presents a comparison of the results of both culturing techniques. Only the genera that occurred in at least 21.7 % (5 of 23) of the positive bone samples with the sequencing method are reported. With 16S rRNA sequencing, we found significantly more anaerobic pathogens (86.9 % vs. 23.1 %, p = 0.001), significantly more Gram-positive bacilli (78.3 % vs. 3.8 %, p < 0.001) and more polymicrobial infections (91.3 % vs. 64.0 %, p = 0.125). Also, greater bacterial diversity was seen both in the Gram-positive cocci and the anaerobes.
No pathogens were identified in 8 out of the 34 bone samples (23.5 %) with conventional culture techniques. Two out of those eight negative samples did not sequence either. The genera that were sequenced in the remaining six and occurred in at least 33.3 % (2 of 6) of the samples are presented in Table 3. Staphylococcus spp. were detected in all of the negative samples, with an average contribution of 21.8 % to the total bacterial population. One of the negative samples was a very low coverage sample (sequence count 3); all three sequences matched with the Staphylococcus spp. sequence derived from the NCBI database. This was the only negative bone sample that had a single genus present.
Conclusions
The primary genus detected in the bone samples of the current study was the Staphylococcus spp., both with conventional culturing techniques and with 16S ribosomal ribonucleic acid (rRNA) gene sequencing. Not only was it detected in 89.6 % (26 of 29) of the sequenced samples, its average contribution to the total bacterial population was the highest of all the genera. This is not a surprising result, since nearly every study reported in the North American and European literature identifies Staphylococcus aureus as the most common pathogen cultured in diabetic foot osteomyelitis (DFO), followed by S. epidermidis [1].
The Corynebacterium spp. was the most prevalent population after Staphylococcus spp. However, the average contribution of this genus to the total bacterial population appears to be much lower. This fastidious organism has been associated with DFO in previous studies that used traditional culturing methods [17, 18]. In a recent study by Dowd et al. [11], the Corynebacterium spp. was even identified as the predominant genus in the individual ecologies of 40 diabetic foot ulcers using a similar sequencing approach. The Staphylococcus spp. was only detected in 13 of the 40 debridement samples. However, the pathogenic role of Corynebacterium spp. in infections is not well understood and the genus is usually considered a contaminant.
Because DFO is not typically exposed to air, especially if peripheral arterial disease is present, anaerobes may play a bigger role than expected. As has been previously reported in studies using pyrosequencing to characterise bacterial diversity in chronic osteomyelitis of the jaw [19], osteomyelitis was not caused by a single pathogen but by diverse bacteria comprising both aerobic and anaerobic species, including unculturable bacteria with conventional culturing methods. Only 3 out of our 29 sequenced bone samples had a single genus of bacteria present, and all three of these samples had low sequence counts, so they may have been contaminants. Our results show that, by using a 16S rRNA sequencing technique, anaerobes were detected in 86.9 % of the positive bone samples (vs. 23.1 % with conventional techniques). The number of anaerobes seems to be largely dependent on the culturing method. In studies by Senneville et al. [6] and Ertugrul et al. [20], obligate anaerobes were identified in only 3 % and 5 % of patients, respectively, after optimising the culturing methods.
All of the patients enrolled in the study were admitted to our hospital with moderate/severe Infectious Diseases Society of America (IDSA) infections that required antibiotics and surgery urgently per IDSA treatment guidelines. Therefore, a limitation of our study design is the high pre-test probability of osteomyelitis and the relatively small number of negative subjects. In addition, we did not have a ‘wash-out’ period with no antibiotic therapy before bone cultures were obtained. While this convention is discussed in the medical literature, there is no direct evidence that it affects the results of either culture technique. However, previous antibiotic therapy may have favoured the results of 16S rRNA sequencing, since pathogens did not need to be viable in order to be detected.
The use of advanced biological molecular technology is of particular interest in DFO, wherein the chronicity of the infection and the adhesion of bacteria in a sessile phenotype may make it difficult to culture these pathogens [21]. The diversity of the bacterial population may contribute to the poor success rates of medical treatment of DFO [22–24]. Studies report prolonged treatment courses with antibiotics in non-surgical cases ranging from 42 to 90 days [25], up to even 40 weeks [26], as well as considerable variation in success (57–70 %) [25]. Culture-specific antibiotic treatment has been reported to provide a higher rate of treatment success compared to empiric therapy [7]. A better understanding of the bacterial diversity in DFO will provide new insights to redirect therapy and might improve clinical outcomes in the future [27].
Abbreviations
- DFI:
-
Diabetic foot infection
- DFO:
-
Diabetic foot osteomyelitis
- IDSA:
-
Infectious Diseases Society of America
- NCBI:
-
National Center for Biotechnology Information
- OTU:
-
Operational taxonomic units
- rRNA:
-
Ribosomal ribonucleic acid
References
Hartemann-Heurtier A, Senneville E (2008) Diabetic foot osteomyelitis. Diabetes Metab 34(2):87–95
Mutluoglu M, Sivrioglu AK, Eroglu M et al (2013) The implications of the presence of osteomyelitis on outcomes of infected diabetic foot wounds. Scand J Infect Dis 45(7):497–503
Citron DM, Goldstein EJ, Merriam CV et al (2007) Bacteriology of moderate-to-severe diabetic foot infections and in vitro activity of antimicrobial agents. J Clin Microbiol 45:2819–2828
Abdulrazak A, Bitar ZI, Al-Shamali AA et al (2005) Bacteriological study of diabetic foot infections. J Diabetes Complicat 19:138–141
Lavery LA, Sariaya M, Ashry H et al (1995) Microbiology of osteomyelitis in diabetic foot infections. J Foot Ankle Surg 34(1):61–64
Senneville E, Melliez H, Beltrand E et al (2006) Culture of percutaneous bone biopsy specimens for diagnosis of diabetic foot osteomyelitis: concordance with ulcer swab cultures. Clin Infect Dis 42(1):57–62
Lázaro-Martínez JL, Aragón-Sánchez J, García-Morales E (2014) Antibiotics versus conservative surgery for treating diabetic foot osteomyelitis: a randomized comparative trial. Diabetes Care 37(3):789–795
Lesens O, Desbiez F, Vidal M et al (2011) Culture of per-wound bone specimens: a simplified approach for the medical management of diabetic foot osteomyelitis. Clin Microbiol Infect 17(2):285–291
American Diabetes Association (1999) Consensus Development Conference on Diabetic Foot Wound Care: 7–8 April 1999, Boston, Massachusetts. American Diabetes Association. Diabetes Care 22:1354–1360
Amann J (1911) Die direkte Zählung der Wasserbakterien mittels des Ultramikroskops. Centralbl Bakteriol 29:381–384
Dowd SE, Wolcott RD, Sun Y et al (2008) Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3:e3326
Lipsky BA, Berendt AR, Cornia PB et al (2012) 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis 54(12):e132–e173
Quince C, Lanzen A, Davenport RJ et al (2011) Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12:38
Edgar RC, Haas BJ, Clemente JC et al (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200
Haas BJ, Gevers D, Earl AM et al (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21(3):494–504
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Bessman AN, Geiger PJ, Canawati H (1992) Prevalence of corynebacteria in diabetic foot infections. Diabetes Care 15:1531–1533
Wheat LJ, Allen SD, Henry M et al (1986) Diabetic foot infections. Bacteriologic analysis. Arch Intern Med 146:1935–1940
Goda A, Maruyama F, Michi Y et al (2014) Analysis of the factors affecting the formation of the microbiome associated with chronic osteomyelitis of the jaw. Clin Microbiol Infect 20:O309–O317
Ertugrul MB, Baktiroglu S, Salman S et al (2006) The diagnosis of osteomyelitis of the foot in diabetes: microbiological examination vs. magnetic resonance imaging and labelled leucocyte scanning. Diabet Med 23(6):649–653
Dowd SE, Sun Y, Secor PR et al (2008) Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8:43
Davis SC, Martinez L, Kirsner R (2006) The diabetic foot: the importance of biofilms and wound bed preparation. Curr Diab Rep 6(6):439–445
Peters EJ, Lipsky BA, Berendt AR et al (2012) A systematic review of the effectiveness of interventions in the management of infection in the diabetic foot. Diabetes Metab Res Rev 28(Suppl 1):142–162
Senneville E, Lombart A, Beltrand E et al (2008) Outcome of diabetic foot osteomyelitis treated nonsurgically: a retrospective cohort study. Diabetes Care 31(4):637–642
Game FL, Jeffcoate WJ (2008) Primarily non-surgical management of osteomyelitis of the foot in diabetes. Diabetologia 51(6):962–967
Embil JM, Rose G, Trepman E et al (2006) Oral antimicrobial therapy for diabetic foot osteomyelitis. Foot Ankle Int 27(10):771–779
Tang YW, Ellis NM, Hopkins MK et al (1998) Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol 36(12):3674–3679
Acknowledgements
Financial support for this work was provided by the NIDDK Diabetic Complications Consortium (DiaComp, www.diacomp.org), grant DK076169. SAVvA collected clinical data and wrote the manuscript. JLF collected data and edited the manuscript. EJGP edited the manuscript. KB was a co-investigator and edited the manuscript. PJK edited the manuscript. LAL collected data and contributed to the manuscript. LAL is the guarantor of this work and takes responsibility for the content of the article. The authors are grateful to Dr. J. Delton Hanson and PathoGenius Laboratories for their contribution and assistance with this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van Asten, S.A.V., La Fontaine, J., Peters, E.J.G. et al. The microbiome of diabetic foot osteomyelitis. Eur J Clin Microbiol Infect Dis 35, 293–298 (2016). https://doi.org/10.1007/s10096-015-2544-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-015-2544-1