
Abstract
Objectives
To investigate the clinical implications of microvascular obstruction (MVO) and intramyocardial haemorrhage (IMH) in acute myocardial infarction (AMI).
Methods
Ninety patients with a first AMI undergoing primary percutaneous coronary intervention (PCI) were studied. T2-weighted, cine and late gadolinium-enhanced cardiovascular magnetic resonance imaging was performed at 5 ± 2 and 103 ± 11 days. Patients were categorised into three groups based on the presence or absence of MVO and IMH.
Results
MVO was observed in 54% and IMH in 43% of patients, and correlated significantly (r = 0.8, p < 0.001). Pre-PCI thrombolysis in myocardial infarction 3 flow was only observed in MVO(−)/IMH(−) patients. Infarct size and impairment of systolic function were largest in MVO(+)/IMH(+) patients (n = 39, 23 ± 9% and 47 ± 7%), smallest in MVO(−)/IMH(−) patients (n = 41, 8 ± 8% and 55 ± 8%) and intermediate in MVO(+)/IMH(−) patients (n = 10, 16 ± 7% and 51 ± 6%, p < 0.001). LVEF increased in all three subgroups at follow-up, but remained intermediate in MVO(+)/IMH(−) and was lowest in MVO(+)/IMH(+) patients. Using random intercept model analysis, only infarct size was an independent predictor for adverse LV remodelling.
Conclusions
Intramyocardial haemorrhage and microvascular obstruction are strongly related. Pre-PCI TIMI 3 flow is less frequently observed in patients with MVO and IMH. Only infarct size was an independent predictor of LV remodelling.
Similar content being viewed by others
Introduction
Coronary occlusion that persists for more than 40 min leads to irreversible myocardial damage that starts in the endocardium and progresses towards the epicardium as a wavefront phenomenon [1]. Although timely reperfusion salvages myocardium and reduces mortality, successful restoration of epicardial artery patency after prolonged occlusion does not always lead to adequate reperfusion at the microvascular level. Reperfused acute myocardial infarction (AMI) is characterised by myocyte swelling due to osmotic overload, intracellular calcium overload, myocyte hypercontraction, microvascular obstruction (MVO) and intramyocardial haemorrhage (IMH) [2, 3].
Both MVO and IMH occur exclusively in myocardium that sustains the most severe ischaemia and are therefore confined to the central portions of infarcted myocardium [3]. As IMH is caused by extravasation of blood into the extravascular space as a consequence of ischaemic microvascular damage, it only occurs in reperfused AMI [4, 5]. IMH does not depend on the magnitude of early reflow but, similar to MVO, on the severity of ischaemia as reflected by a longer duration of coronary occlusion and low collateral flow [6, 7].
Cardiovascular magnetic resonance imaging (CMR) is currently regarded as the reference standard to assess myocardial infarction and allows the detection of MVO and IMH in vivo, using late gadolinium-enhanced (LGE) and T2-weighted (T2W) imaging [8–10]. Previous studies have suggested that MVO might be associated with adverse ventricular remodelling and clinical outcome [9, 11–14]. The clinical significance of IMH is less well defined, and conflicting results have been reported [15, 16].
In this study, we sought to investigate the clinical implications of both MVO and IMH with regard to LV remodelling.
Methods
Study population
We studied 90 consecutive patients (65 men, age 60 ± 11 years) with a first AMI referred for primary percutaneous coronary intervention (PCI) and presentation <12 h after symptom onset. The definition of AMI was based on the recent consensus document including appropriate rise and fall in cardiac biomarkers [17]. Excluded were patients <18 years and those with contraindications for CMR. The institutional review board of our hospital approved the study, and patients were included after written informed consent was obtained.
Before emergency PCI all patients received 500 mg aspirin, 5,000 U heparin and 600 mg clopidogrel. The culprit coronary artery was the LAD in 31%, the RCA in 56% and LCx in 13% of patients; single vessel disease was present in 52%, two vessel disease in 23% and three vessel disease in 25%. All patients received standard post-PCI care, including dual antiplatelet therapy for at least 1 month. Thrombosuction, which was not standard treatment at the time of this study, and downstream administration of intravenous abciximab, intracoronary nitroglycerine and adenosine were left to the discretion of the interventional cardiologist.
Cardiovascular magnetic resonance imaging protocol
Cardiovascular MRI was performed at 5 ± 2 days and 103 ± 11 days after admission. Images were acquired on a 1.5-T MRI system (Intera, Philips Medical Systems, Best, The Netherlands) with a dedicated five-element phased array surface coil. For functional analysis, ECG-gated cine images were obtained in the LV short axis plane covering the entire LV using a segmented balanced steady-state free precession sequence [slice thickness 6 mm, slice gap 4 mm, average repetition time (TR) and echo time (TE) 3.8/1.9 ms, respectively, flip angle 50°, FOV 350 mm, matrix 256 × 256, typically 22–25 phases per cardiac cycle]. Next, a breath-hold, multislice, black blood T2-weighted turbo spin-echo sequence with short inversion time and fat suppression was used (T2 STIR, slice thickness 8 mm, slice gap 2 mm, TR two R-R intervals and TE 100 ms, FOV 350 mm, matrix 512 × 512). LGE CMR was performed 10 min after an intravenous bolus of 0.2 mmol/kg body weight gadolinium-diethylenetriaminepentaacetic acid (Magnevist®, Bayer Schering Pharma, Berlin, Germany) using a breath-hold three-dimensional inversion-recovery gradient-echo sequence [acquired slice thickness 12 mm, reconstructed slice thickness 6 mm, average TR/TE 3.9/2.4 ms, multi-shot (50 profiles/shot) segmented partial echo readout every heart beat (TFE), flip angle 15°, FOV 400 mm, matrix 256 × 256, acquired and reconstructed pixel size 1.56 × 1.56 mm]. The inversion time that optimally suppressed signal of non-infarcted myocardium (typical range 200–280 ms) was determined with a preceding Look-Locker sequence.
CMR image analysis
The CMR images were analysed independently by two observers blinded to clinical data, using commercially available software (CAAS MRV 3.0, Pie Medical Imaging, Maastricht, The Netherlands). The interobserver agreement was excellent (κ value 0.9). Discrepancies were resolved in consensus. Endocardial and epicardial borders were manually traced, excluding the papillary muscles, in the end-diastolic and end-systolic short-axis phases to determine left ventricular end-diastolic volume (LVEDV), end-systolic volume (LVESV), stroke volume (LVSV), ejection fraction (LVEF) and end-diastolic mass (LV mass). These parameters were indexed for body surface area.
Likewise, endocardial and epicardial contours were manually traced on the LGE and T2 images, which were viewed as separate sets. The T2W images of the first CMR examination were used to determine the presence or absence of IMH, defined as a central hypoenhanced area within the hyperintense oedematous area, indicative of the area at risk (AAR). The AAR was quantified by semiautomatic detection using a signal intensity (SI) threshold of >2 SD above a remote non-infarcted region and expressed as a percentage of LV mass. Subendocardial ‘slow flow’ artefacts were carefully excluded, and areas of IMH were included in the AAR analysis by manual adjustment of contours. IMH was manually traced and expressed as a percentage of LV mass.
Infarct size (IS) was quantified on the LGE images of the first CMR using an SI threshold of >5 SD above a remote non-infarcted reference region, including areas of MVO (central hypoenhancement within hyperenhanced area) and expressed as a percentage of LV mass. MVO was quantified by manually tracing the central hypoenhanced area and expressed as a percentage of LV mass.
Statistics
Summary statistics of continuous data with symmetric distribution are expressed as mean ± standard deviation (SD), otherwise as median with interquartile range (IQR). Categorical data are expressed as frequencies with percentages. Univariate group comparisons were conducted with the one-way ANOVA or the non-parametric Kruskal-Wallis test for continuous variables. For categorical variables, chi-square or Fisher’s exact tests were applied. Adjustment for multiple testing was conducted via the false discovery rate (FDR). Detected differences were considered significant when the corrected FDR p value was <0.05. A multiple OLS linear regression model was fitted to quantify the association between IMH and MVO for the MVO(+)/IMH(+) group, adjusting for other clinical variables. To stabilise residual variance, IMH and MVO values, expressed as a proportion of the LV mass, were logarithmically transformed (ln). The final model was used to predict IMH size for the remaining 10 MVO(+)/IMH(−) patients. The estimated IMH values could provide some indication of the possible underlying reasons for the absence of IMH in these patients. A random intercept model analysis was conducted to determine independent predictors of LV remodelling. Statistical analysis was performed with SPSS software (version 17.0 for Windows; SPSS Inc., Chicago, IL).
Results
IMH and MVO
Signal intensity on the T2W and LGE images within the territory of the IRA was increased in all patients. The AAR and IS were 26 ± 12% and 15 ± 11% of LV mass, respectively. MVO was observed in 49 (54%) and IMH in 39 patients (43%), and both were always located subendocardially within the infarct core. IMH was only observed in patients with MVO. A significant correlation was found between MVO and IMH extent (r = 0.8, p < 0.001, Fig. 1), but absolute MVO extent was larger than that of IMH [3.1 ml (IQR 1.6–5.3) vs. 2.2 ml (IQR 1.6–3.5), p = 0.04].

Correlation between IMH and MVO. a Original scale values. b Log transformed values. A good linear correlation is seen between MVO and IMH (r = 0.86, p < 0.001). Open circles indicate observed values and stars indicate predicted values in the MVO(+)/IMH(-) group. Note that most of the predicted values are within the lower range of observed values
Subgroups
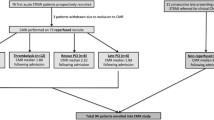
Based on the presence or absence of MVO and IMH, patients were classified into three groups: 41 patients with MVO(−)/IMH(−), 10 with MVO(+)/IMH(−) and 39 with MVO(+)/IMH(+) (Fig. 2). For most of the baseline characteristics, there were no significant differences between groups (Table 1), except for pre-PCI TIMI 3 flow, which was only observed in the MVO(−)/IMH(−) group. There was no difference in the use of glycoprotein IIbIIIa inhibitor between groups.

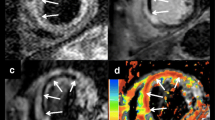
Magnetic resonance image examples from each patient group. Top row (a, b): MVO(−)/IMH(−) patient; middle row (c, d): MVO(+)/IMH(−) patient; bottom row (e, f): MVO(+)/(IMH(+) patient. T2-weighted images are shown on the left (a, c, e) and corresponding late gadolinium-enhanced images on the right (b, d, f). Oedema and infarct border zones are indicated by arrowheads and IMH and MVO by asterisks
The AAR and IS were largest in the MVO(+)/IMH(+) group, intermediate in the MVO(+)/IMH(−) group and smallest in the MVO(−)/IMH(−) group (Table 1). Myocardial salvage, defined as the difference between AAR and IS, was lowest in the MVO(+)/IMH(+) group, intermediate in the MVO(+)/IMH(−) group and highest in the MVO(−)/IMH(−) group (28 ± 24%, 40 ± 25% and 54 ± 32%, respectively, p < 0.001, Table 1 and Fig. 3).

Area at risk (AAR) and infarct size (IS). AAR (grey bars) and IS (white bars) represented as percentages of LV mass for each patient group. The MVO(+)/IMH(−) group represents an intermediate group. Myocardial salvage is the difference between AAR and IS
The MVO extent tended to be larger in the MVO(+)/IMH(+) patients compared with the MVO(+)/IMH(−) patients [1.7% (IQR 1.0–3.3%) vs. 0.8% (IQR 0.4–1.9%) of LV mass, respectively, p = 0.07].
Predicted IMH size in patients with MVO but without IMH
The IMH in the MVO(+)/IMH(−) patients could have been missed as a result of the limits of spatial resolution of T2W CMR. To investigate this, predicted IMH size values for MVO(+)/IMH(−) were calculated based on the following model derived from the MVO(+)/IMH(+) group data:
All predictors in the above model were statistically significant. Observed and predicted IMH values are displayed in Fig. 1a and b. Predicted values are marked as stars. In either case it is evident that only two patients had sufficiently small predicted IMH outside the observed value range in the other patients. Theoretically, these two values could have been missed by T2W CMR. Conversely, several of the predicted values were still of sufficient size, at least comparable to that of the observed counterparts, and as such they could have been detected by CMR.
Functional parameters and LV remodelling
The LVEDVi and LVESVi decreased from baseline to follow-up and were highest in the MVO(+)/MVO(+) group, intermediate in the MVO(+)/IMH(−) group and lowest in the MVO(−)/IMH(−) group at both time points (Table 2). Likewise, LVEF improved in all three groups, but remained lowest in the MVO(+)/MVO(+) group, intermediate in the MVO(+)/IMH(−) group and highest in the MVO(−)/IMH(−) group in the univariate comparisons (Fig. 4).

Left ventricular ejection fraction (LVEF) at baseline and follow-up. LVEF significantly increased in all three groups from baseline (grey bars) to follow-up CMR (white bars), but remained intermediate in the MVO(+)/IMH(−) group
Table 3 displays the fixed effect parameter estimates of the random intercept model. Subjects were taken as random effect, whereas time and MVO/IMH groups were taken as fixed effect factors adjusted for IS. LVEF significantly increased over time (for all groups), showing a significant negative association with IS. Note that the presence of IMH or MVO had no significant independent predictive value for LVEF once adjusted for IS.
Discussion
The findings of our study can be summarised as follows: (1) MVO and IMH are frequently observed in reperfused AMI using T2W and LGE CMR, (2) IMH and MVO are strongly related phenomena, (3) patients with MVO and IMH have pre-PCI TIMI 3 flow less often, and (4) infarct size, once adjusted for MVO and IMH presence, remained the sole independent predictor of LV remodelling.
T2W CMR is capable of detecting myocardial oedema as regions of increased signal intensity due to prolongation of T2 relaxation time as a consequences of increased tissue free water content [18]. IMH is thought to be caused by leakage of blood out of severely injured microvasculature into the interstitium that can be depicted as a central area of low signal intensity (SI) within the oedematous area on T2W CMR. This low SI is explained by local field heterogeneities due to the paramagnetic effects of blood degradation products such as deoxyhaemoglobin, which shorten the T2 relaxation time [10]. In two case reports, Basso et al. demonstrated a good correlation among the location, spatial extent, and shape of IMH on CMR and histopathology [8]. Because of its excellent spatial resolution and high contrast, LGE CMR accurately assesses the extent of infarction and MVO [19]. Combined with T2W CMR, it is now possible to gain novel insights into the pathophysiology of IMH and MVO in vivo and evaluate their clinical significance.
Previous studies have shown that MVO infarcts show a lack of regional and global functional recovery compared with non-MVO infarcts; however, it remains unclear whether MVO provides independent prognostic information [11, 12]. Despite the strong relation of MVO with IS, some studies indicated that MVO is an important determinant of LV recovery and prognosis, even after controlling for IS, whereas others have reported that MVO loses its predictive power after adjusting for IS [13, 14, 20, 21]. Persistence of MVO, but not MVO that disappeared at 1 week after infarction, was associated with attenuated infarct healing and subsequent adverse remodelling, indicating that the timing of MVO imaging is important [22]. In that study and similar to our findings, IS remained the major determinant of LV remodelling in a multivariate model.
Our clinical observation that MVO and IMH occur in the central portions of the infarct and not in patients with pre-PCI TIMI 3 flow is in line with experimental studies showing the association of IMH with markedly depressed flow before reperfusion and severe ischaemia [5]. Our finding that IMH was also observed in 10% of patients with post-PCI TIMI flow <3 supports experimental observations that IMH does not depend on the magnitude of early reflow [6, 23]. Conflicting results on the clinical significance of IMH have been reported. Our data are in contrast to those of Ganame et al., who found that LVEF did not improve in patients with haemorrhagic infarcts as opposed to patients with non-haemorrhagic infarcts [16]. IMH and infarct size were the strongest independent predictors of adverse LV remodelling in that study. Our results are in accordance with those from Beek et al., who showed that IMH did not have prognostic significance beyond the presence of MVO [15].
The underlying pathophysiological mechanisms of MVO and IMH are complex and remain only partially understood. Irreversible ischaemic myocardial cell injury progresses as a ‘wave front’ from the endocardium to the epicardium, with endothelial cell injury occurring after myocardial cell injury [1, 3]. After varying periods of ischaemia, sequential changes of the microvasculature occur. Endothelial protrusion by cell swelling together with capillary plugging by neutrophils, red blood cells and platelets and swelling of surrounding myocytes compressing capillaries may all cause MVO. As ischaemic injury progresses, endothelial cell death causes loss of capillary integrity with resultant IMH in cases of myocardial reperfusion. Our data show that MVO and IMH are strongly related phenomena and suggest that IMH is a sign of severely injured microvasculature that easily leaks blood into the interstitial space. Furthermore, our results suggest that microvascular injury is predominantly a reflection of extensive myocardial necrosis rather than a separate entity identifying patients at higher risk.
Despite having MVO, IMH was absent in ten patients in our study. We postulate that, for some patients in the MVO(+)/IMH(−) group, the ischaemic threshold for IMH may not have been reached yet. This could explain the discrepancy between the predicted IMH values and the negative findings at CMR. Lending support to this concept was the finding that all measured parameters, including MVO size, were in between the average values of the two other groups. It is therefore likely that the groups reflect a gradation of the severity of ischaemic damage.
Conclusions
Microvascular obstruction and intramyocardial haemorrhage are frequently observed in reperfused AMI using T2W and LGE CMR, and are strongly related phenomena. Patients with MVO and IMH have pre-PCI TIMI 3 flow less often, suggesting more severe ischaemia before reperfusion. Only infarct size, but not the presence of IMH or MVO, was an independent predictor for LVEF. This suggests that MVO and IMH are not just separate entities, but rather ischaemia-dependent consequences of severely injured microvasculature in larger infarcts.
References
Reimer KA, Lowe JE, Rasmussen MM et al (1977) The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 56:786–794
Bresnahan GF, Roberts R, Shell WE et al (1974) Deleterious effects due to hemorrhage after myocardial reperfusion. Am J Cardiol 33:82–86
Kloner RA, Ganote CE, Jennings RB et al (1974) The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest 54:1496–1508
Basso C, Thiene G et al (2006) The pathophysiology of myocardial reperfusion: a pathologist's perspective. Heart 92:1559–1562
Fishbein MC, J Y-Rit, Lando U et al (1980) The relationship of vascular injury and myocardial hemorrhage to necrosis after reperfusion. Circulation 62:1274–1279
Garcia-Dorado D, Theroux P, Solares J et al (1990) Determinants of hemorrhagic infarcts. Histologic observations from experiments involving coronary occlusion, coronary reperfusion, and reocclusion. Am J Pathol 137:301–311
Higginson LA, White F, Heggtveit HA et al (1982) Determinants of myocardial hemorrhage after coronary reperfusion in the anesthetized dog. Circulation 65:62–69
Basso C, Corbetti F, Silva C et al (2007) Morphologic validation of reperfused hemorrhagic myocardial infarction by cardiovascular magnetic resonance. Am J Cardiol 100:1322–1327
Hombach V, Grebe O, Merkle N et al (2005) Sequelae of acute myocardial infarction regarding cardiac structure and function and their prognostic significance as assessed by magnetic resonance imaging. Eur Heart J 26:549–557
Lotan CS, Bouchard A, Cranney GB et al (1992) Assessment of postreperfusion myocardial hemorrhage using proton NMR imaging at 1.5 T. Circulation 86:1018–1025
Baks T, van Geuns RJ, Biagini E et al (2006) Effects of primary angioplasty for acute myocardial infarction on early and late infarct size and left ventricular wall characteristics. J Am Coll Cardiol 47:40–44
Bogaert J, Kalantzi M, Rademakers FE et al (2007) Determinants and impact of microvascular obstruction in successfully reperfused ST-segment elevation myocardial infarction. Assessment by magnetic resonance imaging. Eur Radiol 17:2572–2580
Nijveldt R, Beek AM, Hirsch A et al (2008) Functional recovery after acute myocardial infarction: comparison between angiography, electrocardiography, and cardiovascular magnetic resonance measures of microvascular injury. J Am Coll Cardiol 52:181–189
Wu KC, Zerhouni EA, Judd RM et al (1998) Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 97:765–772
Beek AM, Nijveldt R, van Rossum AC et al (2009) Intramyocardial hemorrhage and microvascular obstruction after primary percutaneous coronary intervention. Int J Cardiovasc Imaging. doi:10.1007/s10554-009-9499-1
Ganame J, Messalli G, Dymarkowski S et al (2009) Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur Heart J 30:1440–1449
Thygesen K, Alpert JS, White HD et al (2007) Universal definition of myocardial infarction. Circulation 116:2634–2653
Simonetti OP, Finn JP, White RD et al (1996) “Black blood” T2-weighted inversion-recovery MR imaging of the heart. Radiology 199:49–57
Kim RJ, Fieno DS, Parrish TB et al (1999) Relationship of MRI delayed contrast enhancement to irreversible injury, infarct age, and contractile function. Circulation 100:1992–2002
Shapiro MD, Nieman K, Nasir K et al (2007) Utility of cardiovascular magnetic resonance to predict left ventricular recovery after primary percutaneous coronary intervention for patients presenting with acute ST-segment elevation myocardial infarction. Am J Cardiol 100:211–216
Tarantini G, Razzolini R, Cacciavillani L et al (2006) Influence of transmurality, infarct size, and severe microvascular obstruction on left ventricular remodeling and function after primary coronary angioplasty. Am J Cardiol 98:1033–1040
Orn S, Manhenke C, Greve OJ et al (2009) Microvascular obstruction is a major determinant of infarct healing and subsequent left ventricular remodelling following primary percutaneous coronary intervention. Eur Heart J 30:1978–1985
Roberts CS, Schoen FJ, Kloner RA et al (1983) Effect of coronary reperfusion on myocardial hemorrhage and infarct healing. Am J Cardiol 52:610–614
Disclosures/conflict of interests
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bekkers, S.C.A.M., Smulders, M.W., Passos, V.L. et al. Clinical implications of microvascular obstruction and intramyocardial haemorrhage in acute myocardial infarction using cardiovascular magnetic resonance imaging. Eur Radiol 20, 2572–2578 (2010). https://doi.org/10.1007/s00330-010-1849-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00330-010-1849-9