
Abstract
The role of islet amyloidosis in the onset and progression of Type 2 diabetes remains obscure. Islet amyloid polypeptide is a 37 amino-acid, beta-cell peptide which is co-stored and co-released with insulin. Human islet amyloid polypeptide refolds to a β-conformation and oligomerises to form insoluble fibrils; proline substitutions in rodent islet amyloid polypeptide prevent this molecular transition. Pro-islet amyloid polypeptide (67 amino acids in man) is processed in secretory granules. Refolding of islet amyloid polypeptide may be prevented by intragranular heterodimer formation with insulin (but not proinsulin). Diabetes-associated abnormal proinsulin processing could contribute to de-stabilisation of granular islet amyloid polypeptide. Increased pro-islet amyloid polypeptide secretion as a consequence of islet dysfunction could promote fibrillogenesis; the propeptide forms fibrils and binds to basement membrane glycosamino-glycans. Islet amyloid polypeptide gene polymorphisms are not universally associated with Type 2 diabetes. Transgenic mice expressing human islet amyloid polypeptide gene have increased islet amyloid polypeptide concentrations but develop islet amyloid only against a background of obesity and/or high fat diet. In transgenic mice, obese monkeys and cats, initially small perivascular deposits progressively increase to occupy 80% islet mass; the severity of amyloidosis in animal models is related to the onset of hyperglycaemia, suggesting that islet amyloid and the associated destruction of islet cells cause diabetes. In human diabetes, islet amyloid can affect less than 1% or up to 80% of islets indicating that islet amyloidosis largely results from diabetes-related pathologies and is not an aetiological factor for hyperglycaemia. However, the associated progressive beta-cell destruction leads to severe islet dysfunction and insulin requirement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Amyloid deposition in pancreatic islets is one of the most common pathological features of Type 2 diabetes (T2DM) being found in at least one islet at post-mortem in more than 90% of Type 2 diabetic subjects [1, 2, 3]; islet amyloid is not just a feature of ageing since it is found only in a small number of (<15%) elderly (>60 years), apparently non-diabetic individuals. This pathological feature was first identified by a study in a pancreas from a diabetic patient in 1900 [4]. Later pathologists determined that islet hyaline plaques are a feature of elderly (but not young) subjects with diabetes [5] and that this amorphous proteinaceous material (which was originally described as hyaline), had tinctorial properties of amyloid [6], staining with Congo red or thioflavin S (Fig. 1a,b). Causative factors for conversion of the normally soluble component peptide, islet amyloid polypeptide (IAPP, amylin) to insoluble fibrils remains largely unknown. Amyloidogenic peptides change their conformation to form β-sheets and fibrils; Aβ peptide which forms cerebral plaques in Alzheimer’s disease (AD) [7] and is of similar size to IAPP, shows similar conformational changes. However, any common factors which could link AD with T2DM remain to be determined [8, 9].

Islet amyloid deposits fluorescently labelled with Thioflavine S in islets of two subjects with Type 2 diabetes to illustrate that the degree of severity of the deposits (% islet occupied) does not correlate with the duration or severity of the diabetic symptoms. (a) Pancreatic islet from a subject (aged 86 years) who died after 18 years of Type 2 diabetes, treated with oral agents; quantitation data indicated less than 2% islets contained amyloid with less than 3% islet area replaced with amyloid. Amyloid was present as thin perivascular deposits adjacent to the capillary surrounding the islet and those penetrating the core with limited accumulation within the islet. (b) Subject aged 76 years died after 17 years of diabetes treated with oral agents; 52% of islets were affected with amyloid occupying mean of 20% islet space. Perivascular fibrils and extensive deposits within the islet were present associated with a reduction of beta-cell population. Quantitation data derived from studies reported in ref [3]. Scale bars=20 µm
The relationship of islet amyloid to the onset and progression of T2DM in man is largely unknown; formation of amyloid cannot be directly related to the pathophysiology of diabetes since the deposits are undetectable in vivo and animal models of T2DM do not include all features of the disease process. Extensive islet amyloid deposition identified at post mortem has been correlated with reduced islet function and loss of islet beta cells in animal models [10] and with severe islet dysfunction associated with the need for insulin replacement therapy in humans [1, 11]. Amyloid fibrils formed from synthetic IAPP [12, 13] or Aβ peptides [7] are cytotoxic which has significance for islet dysfunction in diabetes.
This review describes current information on the factors that affect human IAPP (hIAPP) fibril formation in vitro and relates these findings to fibril formation and accumulation of deposits in biological systems. The relationship of fibril formation to islet function in man and in cellular and animal models is discussed to assess the role of amyloidosis in the different stages of T2DM in man.
What is IAPP and islet amyloid?
Islet amyloid polypeptide, IAPP, amylin, is a 37 amino acid peptide which is co-stored with insulin in beta-cell secretory granules and co-secreted in response to beta-cell secretagogues [14, 15, 16]. IAPP is derived from a larger precursor peptide, proIAPP, (67 amino acids in man) and proteolytically cleaved at the N- and C-terminal junctions by prohomone convertase 1/3 and 2 within the beta-cell secretory granule as the granule matures (Fig. 2) [17, 18, 19, 20]. The normally soluble peptide is found in the circulation at 5–15 pmol/l concentration in man and like C-peptide (but not insulin) it is excreted by the kidney; the relationship of circulating insulin-like molecules to IAPP would therefore be more accurately made with C-peptide than insulin [21].

Amino acid sequences of proIAPP illustrated in single letter code. Three residues are missing in human, monkey, cat and dog when aligned with other sequences. Porcine IAPP is missing one amino acid. Amino acids common to the human sequence are indicated by (-). Asterisks (*) represent amino acids not yet determined. : represent residues missing when sequences are aligned. Proline residues in IAPP 20–29 are present in rat and mouse IAPP which prevent folding to form fibrils. Only partial sequences are available for rabbit, hare, cougar, and pig. G-pig guinea-pig
IAPP has been identified in all species in which it has been examined [21]. The primary structure of IAPP is well conserved although there are some important species–specific substitutions which have implications for amyloid formation (Fig. 2). Diabetes-associated amyloid is found in non-human primates [23, 24] and cats [25] but not in rodents. Proline substitutions in the region IAPP 24–29 are responsible for the lack of fibril formation in rats and mice and some other species [26, 27, 28, 29]. Islet amyloid is not formed in Type 1 diabetic subjects where beta cells (and therefore insulin and IAPP) are absent. IAPP has some homologies to calcitonin gene-related peptide (CGRP) [30] and shares some actions and receptor binding affinities with the calcitonin gene family of peptides [31, 32, 33]. Although a multitude of physiological roles have been attributed to IAPP since its identification in 1987 [34, 35], it remains a peptide without a clearly identified function in vivo [36]; moderately improved glucose intolerance was detected in the IAPP knock-out mouse [37, 38] and Type 1 diabetic subjects do not have pathophysiological features which could be attributed directly to IAPP deficiency.
The extent of islet amyloid deposition in T2DM is very variable; not only is the number of islets affected (prevalence) very heterogeneous (<1%–80%) but also the amount of amyloid per islet (severity) is not uniform [39]. Small deposits are located adjacent to the basement membrane of islet capillaries (Fig. 1a, Fig. 3). Islets in the head of the pancreas are less affected than those in the remainder of the organ due to the reduced number of beta cells in islets derived from the ventral pancreatic primordium [40].

Perivascular islet amyloid deposits examined by electron microscopy in (a) a human diabetic subject, (b) a glucose-intolerant Macaca mulatta monkey and (c) an old (aged 18 months), non-diabetic, non-obese, transgenic mouse expressing the gene for human IAPP. (a) Amyloid deposits are situated between an islet capillary and the beta cell; the amyloid fibrils lie in invaginations of the beta-cell membrane (arrows). This could disrupt membrane signalling of glucose uptake and insulin secretion by interference with membrane fluidity. (b) perivascular amyloid surrounds this profile of the capillary and appears to be more extensive adjacent to the beta cell than the α-cell suggesting an association with beta-cell pathology. (c) Amyloid deposits were very infrequent in this old non-diabetic transgenic mouse. The deposit in this profile is more extensive adjacent to the beta cell than the α cell. Amy, amyloid deposits; c, capillary; β, beta cell; α, α cell. *, basement membrane. Scale bars (a) 2.0 µm, (b) 5.0 µm, (c) 1.0 µm
Islet amyloid deposits contain a variety of components that are common to all forms of amyloidosis; serum amyloid P component [41], apolipoprotein E (ApoE) and heparan sulphate proteoglycans [42, 43, 44] are present in addition to the proteinaceous fibrils; some of these molecules may play a role in the initiation or stabilisation of the deposits. All amyloid fibrils have similar structural features independently of the component protein [45]. In the case of islet amyloid, the normally soluble secreted peptide assembles into a rigid linear structure with each monomer held together via hydrogen bonds; it is possible that an amorphous precipitate of monomers/small oligomers forms as seen in vitro (Fig. 4a) which provides the local conditions for fibril assembly [46]. Fibrils start as small units, protofibrils, consisting of a relatively small number of oligomers (Fig. 4b); these further assemble to form a filament which is 4 to 8 nm in diameter and unbranching (Fig. 4c). Fibrils (10–30 nm diameter and of variable length) are usually composed of 2 to 5 protofilaments wound together (Fig. 4d,e) which form the foundation of extracellular deposits (Fig. 4f). Most amyloid fibrils are found outside the cells in vivo but considerable controversy exists concerning the site of initiation of islet amyloidosis and the possibility of fibril assembly inside the cell [47, 48]: fibril-like accumulations of IAPP with tinctorial properties of amyloid have been identified at intracellular sites in beta cells of human insulinomas and of islets from transgenic mice overexpressing the gene for hIAPP (hIAPP TM) [22, 49].

Different stages in the assembly of fibrils formed from human synthetic IAPP. (a) Human IAPP precipitates from solution and forms amorphous aggregates visible with transmission electron microscopy; this hydrophobic environment allows structural refolding and formation of oligomers and protofibrils. (b) Protofibrils (arrows) consist of small oligomers of IAPP which are illustrated using platinum carbon shadowing and electron microscopy. (c) Further assembly of protofibrils result in protofilaments which are long and unbranching but are not stable alone. (d) These associate laterally, often by helical winding, to form fibrils consisting of three or more protofilaments. (e) Fibrils formed from synthetic peptides also assemble into loose meshworks of fibrils. (f) Ex-vivo fibrils extracted from the pancreas of a diabetic subject show little evidence of complex helical or higher order fibril assembly. Scale bars, 200 nm
What are the causative factors for amyloid fibril formation in Type 2 diabetes?
What normally prevents IAPP conversion into fibrils?
Synthetic hIAPP in buffer solutions is rapidly converted to β-sheet and fibrils [45, 49, 50]. In the secretory granule, IAPP is at a high concentration; this is approximately 4 mmol/l based on a ratio of 1:10 with insulin in pancreatic extracts [52] and an estimated concentration of granular insulin of 40 mmol/l [53]; at this concentration, synthetic hIAPP would be rapidly converted into fibrils. This suggests that the peptide must be stabilised to prevent oligomerisation and fibril formation in vivo. Many laboratories have shown that insulin will inhibit IAPP fibril formation in vitro [54, 55]; IAPP and insulin (but not proinsulin) form heteromolecular complexes in vitro [56] suggesting that insulin stabilises hIAPP in the beta-cell granule. Proinsulin does not bind to IAPP or have a stabilizing effect [56] and, therefore, any process that results in inefficient proinsulin processing could affect IAPP fibril formation in T2DM.
Environmental destabilisation
IAPP fibril formation results from a change in the normal conformational state of the molecule to a new, aggregation-prone conformation [57, 58, 59]. The ‘environmental’ factors that can destabilise a protein conformation include pH, chemical modification, salt concentrations, natural ligands, glycation, racemisation, deamidation, oxidation, isomerisation and lipidoxidation of the peptide. None of these modifications of IAPP have been assessed in vivo and, therefore, their potential association with amyloid deposition is circumstantial. Experiments of IAPP model peptides show that deamidation can dramatically accelerate amyloid formation [60]. When the secretory granule is released, the granule milieu of the normally acid pH and high calcium concentration is changed to neutral and lower calcium of the extracellular space and the insulin crystal dissolves. Environmental changes associated with exocytosis do not promote IAPP molecular aggregation in vitro [58].
Genetically determined structural changes- mutations in the IAPP gene
No genetically-determined aberrant IAPP sequence has been found in the majority of populations with T2DM [61, 62]. A missense heterozygous substitution in hIAPP at position 20 of a serine residue for a glycine has been identified in Japanese and Chinese studies [63]. However the prevalence of the mutation in T2DM is low, although significant, in both Japanese (2.6% in T2DM vs 0.8% in non-diabetic group, p<0.0007) [64] and Chinese cohorts (2.0–2.8% in T2DM; 0–0.5% in controls, p<0.05) [65] but not found in large cohorts of Caucasians [66]. The S20G mutation is associated with only moderate alterations in insulin secretion in the small number of patients examined so far and, therefore, its role in T2DM is unclear [67]. However, synthetic S20G hIAPP forms amyloid more readily than wild-type IAPP [68] and it exerts greater biological cytotoxicity [69] indicating that small changes in structure can affect the refolding process. A mutation in the IAPP gene promoter has been identified and associated with T2DM in a Spanish population [70] which suggests that regulation of gene expression could be important.
Aberrant prohormone processing
The role of inappropriate processing of proIAPP in T2DM remains high as a candidate factor for fibrillogenesis. In T2DM [71] and in patients with insulinoma (both conditions associated with amyloid) there is increased secretion of incompletely processed proinsulin, [72, 73]. Islet studies in vitro indicate increased production of N-terminal intact proIAPP under stimulated secretory conditions [74] and immunoreactivity for the N- terminal peptide of proIAPP has been identified in islet amyloid deposits in insulinomas and T2DM [75]. N-terminal intact proIAPP has been identified in a PC2 knockout, hIAPP transgenic mouse model [76]. This suggests that, not only is inappropriately processed proinsulin secreted under these conditions, but also that proIAPP secreted in T2DM is incorporated into deposits.
An increase in production of proIAPP-like molecules could affect IAPP aggregation by several mechanisms. Firstly, proIAPP has been shown to form amyloid-like fibrils in vitro although with less avidity than IAPP [77]. Secondly, it has been shown that a synthetic construct of proIAPP with the N-terminal cleavage fragment intact, readily binds to heparan sulphate but IAPP does not [78]. Since heparan sulphate is a component of the glycosaminoglycans (GAGs) of the islet capillary basement membrane (where amyloid first forms in vivo), secreted proIAPP could accumulate in the pericapillary space and either form fibrils itself or act as a nidus or template for subsequent IAPP accumulation. Alternatively, GAGs produced by the beta cell could act as binding sites for IAPP [78]. Thirdly, insulin (but not proinsulin) binds to IAPP and prevents fibril formation [56]; therefore, aberrant proinsulin processing and increased concentration in the granule could lead to destabilisation of IAPP and/or proIAPP which could result in fibril formation on granule exocytosis. Fibril formation as a result of peptide misprocessing would then result from, rather than cause, beta-cell dysfunction.
Advanced glycation end products of insulin and IAPP
Protein glycation is a feature of T2DM and the formation of advanced glycation end products (AGEs) is demonstrable in proteins with low turnover rates (e.g. haemoglobin, collagen) [80]. The use of fructose as a sweetener could exaggerate the problem because the chemical reaction of fructose with proteins is faster than the analogous reaction with glucose. Glycated insulin has been identified in models of diabetes [81] but the existence of glycated IAPP in the circulation is unknown. Whilst glycation of established amyloid deposits is likely in diabetes, glycation of molecules which are rapidly turned over once secreted into the circulation such as insulin and IAPP would need to occur within the cell. Glycation of IAPP in vitro occurs at the α-amino or ε-amino group of Lys1 and guanidine group of Arg11 and this has been proposed to accelerate fibril formation [82]. Glycation of proteins in diabetes could affect cellular viability through binding to the so-called Receptor for Advanced Glycation End products or RAGE. Monomeric non-glycated hIAPP does not bind to this receptor but fibrillar IAPP (and amyloid fibrils composed of other proteins) have been shown to interact [84]. If glycated monomeric IAPP binds to the RAGE receptor this could act as a “nidus” for fibril formation and/or induce apoptosis but there is no evidence from in vivo studies for this mechanism.
IAPP concentration
As with other amyloid diseases, the extent of islet amyloid formation is related to the production and local concentrations of IAPP; these are dependent upon secretion, clearance from the islet spaces or by degradation. No correlation has been observed between an increase in circulating IAPP concentrations and either glucose intolerance or T2DM in man other than that which would be expected as a result of increased insulin resistance [10, 16, 84]. In addition, insulin and IAPP are co-ordinately regulated in terms of gene expression [85] and circulating concentrations in glucose intolerance and diabetes in man [16, 84, 86, 87]; disassociation of the two beta-cell products does however occur during treatment with steroids [97, 98, 99] when expression of insulin is reduced and IAPP enhanced.
Transgenic mice overexpressing the gene for human IAPP (hIAPP TM) have increased circulating IAPP which can be up to 30 times higher than the endogenous mouse IAPP [49, 91, 92, 93, 94]; these are not necessarily associated with amyloidosis suggesting that increased concentration is not adequate alone to induce changes in peptide conformation [95, 96]. Amyloid deposition in obese or fat-fed hIAPP TM is more frequent in males [96, 97], is reversed by oestrogen treatment in obese male animals [98] and is increased in hIAPP females by oophorectomy despite no change in circulating concentrations of IAPP [99]. No such clear sex differences in diabetes susceptibility are seen in humans or in cats and monkeys. The fact that increased dietary fat promotes islet amyloidosis in islets of transgenic mice in vivo [97] suggests that the role of lipids in the genesis of amyloid fibrils is important in addition to the increased insulin resistance and beta-cell secretion observed in obesity. In vitro, fatty acids promote IAPP fibrillogenesis of synthetic hIAPP and biosynthetic IAPP in hIAPP mouse islets [100, 101] and the degree of amyloidosis in hIAPP TM in vivo was shown to be proportional to the percentage of fat in the diet [102]. However, despite a three-fold increase in cholesterol in ApoE knockout mice expressing hIAPP there was no increase in the degree of islet amyloidosis [103]. Amyloid was not associated with obesity in non-diabetic Pima Indians or Caucasian subjects [3, 104]. If obesity and associated increases in circulating non-esterified fatty acids and other lipid fractions in diabetic man influence fibril formation, islet amyloidosis could be considered to be a complication of diabetes-related obesity.
Increased local concentrations of secreted peptides are also influenced by clearance. IAPP fibrils form between beta cells in islets from hIAPP TM maintained in culture (Fig. 5) [49, 105]; the degree of extracellular fibril formation in these islets was increased by beta-cell secretagogues suggesting that a combination of increased secretion and decreased clearance in the poorly-perfused, extracellular spaces contribute to fibrillogenesis [105]. Disturbances in the islet vasculature and blood flow are associated with impaired glucose tolerance in animal models of diabetes [106]; decreased cardiovascular perfusion of islets in diabetes could account for decreased IAPP clearance. In a similar way, IAPP (and proIAPP) clearance could be reduced by binding to heparan sulphate or a RAGE receptor on the capillary basement membrane.

Biosynthetic fibrils forming in vitro in an islet isolated from a TM-expressing the hIAPP gene examined by electron microscopy. Fibrils formed in the extracellular space between beta cells in islets cultured in 16.7 mmol/l glucose. The cell membrane showed characteristic invaginations filled with fibrils (arrows) as seen in man. Immunogold labelling for IAPP showed immunoreactivity in fibrils, insulin granules (I) and lysosomes (Lyso, arrow). No evidence of fibril-induced cytotoxicity was present in the cells adjacent to the biosynthetic fibrils. Scale bar 200 nm
Under normal conditions, beta-cell granules including intracellular IAPP and insulin which are not targeted for secretion, are degraded in lysosomes [107] and human IAPP accumulates in lysosomes in islets from man and hIAPP transgenic mice [108, 109]; however, there was no evidence of IAPP fibril formation in lysosomes indicating that an increased concentration is not sufficient for intracellular fibril formation at this site. A proposal has been made that all amyloid deposits are being degraded continuously [110], possibly by removal by macrophages, and that accumulation of deposits could result from a deficiency in the factors responsible for degradation and/or turnover [111]. Although IAPP fibrils have been detected in pancreatic macrophages [112], amyloid deposition is not associated with increased macrophage density [113]. The many amyloid-associated factors including the amyloid P component [111] and glycosaminoglycans have been proposed to prevent recognition of deposits for phagocytosis by macrophages and thus reduce clearance of deposits [111].
The relationship of islet amyloid to the pathophysiology of Type 2 diabetes; a complication or causative factor?
Islet amyloid and glucose intolerance
The relationship of islet amyloid to hyperglycaemia, insulin resistance and beta-cell dysfunction in vivo in man is difficult to establish since there is no quantifiable clinical feature that can be related to the deposits and pancreatic biopsies are ethically unacceptable. Spontaneously developing diabetes in cats and monkeys is associated with progressively increasing islet amyloidosis [23, 24, 25]. These models of diabetes have a similar physiological syndrome to that seen in man, including older age of onset, obesity, impaired glucose tolerance progressing to hyperglycaemia and insulin dependence. Longitudinal and cross-sectional studies in these animals have shown that, as in man, amyloid deposition commences as fine deposits adjacent to islet capillaries (Fig. 1b). In these non-human species and in transgenic hIAPP, fat-fed mice [114], initial fibril formation accompanies or precedes obesity and/or glucose intolerance and, at this stage, only a few islets are affected (low prevalence) with no significant change in islet-cell mass [114]. However, very small perivascular deposits, whilst not apparently cytotoxic, could compromise transfer of nutrients and insulin across the pericapillary space in affected islets and contribute to glucose intolerance in these models. As the deposition progresses more islets become affected (increased islet prevalence) and when most islets are affected there is a substantial increase in severity (larger deposits in islets); careful measurements in transgenic mice have shown that a prevalence of 80% was associated with 1.5% of islet space occupied by amyloid [114]. Using similar calculations, a similar progressive relationship between prevalence and severity has been identified in Macaca mulatta islets (Fig. 6); the severity was generally less than 10% in glucose intolerant animals and severity increased when almost 100% of islets were affected. All diabetic animals had extensive amyloidosis suggesting that the islet pathology was a causative factor for onset of hyperglycaemia [23] (Fig. 6). Similarly, hIAPP TM possessing moderate islet replacement with amyloid (~10% islet mass filled with amyloid and 100% islets affected) did not exhibit fasting hyperglycaemia but had slightly impaired glucose stimulated insulin secretory response [114] implicating islet amyloid (and the associated destruction of islet cells) directly with impaired insulin secretion. The later stages in mice, when there is extensive replacement of cells, is similar to that observed in man when insulin therapy is required [1, 2, 11]. The role of amyloid in the onset and progression of diabetes in man is more complex. In patients with diabetes of long duration, the degree of islet amyloidosis at post-mortem can vary from a low prevalence (<1% islets affected with small perivascular deposits) to up to 90% islets affected and a high degree of amyloid replacement of islet cells (up to 80% islet mass occupied with amyloid) [3] (Fig. 1a,b). This indicates that islet amyloid and cellular replacement is not the sole precipitating factor for islet dysfunction and diabetes in man. The requirement for insulin therapy associated with increased amyloid severity occurs subsequent to diagnosis (or not at all in many patients); this degree of progressive amyloidosis is therefore more likely to result from, rather than cause, islet dysfunction (Fig. 7). However, there is no relationship of duration of diabetes to the degree of islet amyloidosis in man which reflects the heterogeneity of the disease. Clinical diagnosis of diabetes can occur months or years subseqent to onset of the syndrome. Identification of the different predisposing factors that can contribute to the aetiology and progression of islet dysfunction (one of which could be islet amyloid), is still largely impossible. Whilst animal models provide excellent longitudinal evidence for the progressive relationship between islet dysfunction and amyloidosis, the deposits seem to precipitate diabetes in these models which is a different scenario to that found in man.

The relationship between islet amyloid prevalence (% islets affected) and amyloid severity (% islet area occupied) in Macaca mulatta monkeys. Cross-sectional data obtained from animals killed at different stages of the diabetes syndrome; no amyloid was found in young, non-diabetic, normal weight animals n=3; low prevalence and severity of deposits were found in 3/9 obese, normoglycaemic monkeys (●); variable degrees of amyloidosis was present in 4/6 obese insulin resistant, glucose intolerant animals (▲); amyloid was present in 8/8 diabetic animals and severity was greater than 70% in 7/8 animals with 100% prevalence (■). Amyloid severity plotted as logarithm for illustrative purposes. Data derived from studies reported in reference [23]

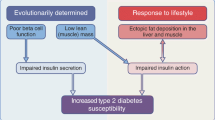
Schematic representation of the potential relationship of islet dysfunction, islet amyloid and beta-cell destruction in the pathophysiology of Type 2 diabetes. Environmental factors, inactivity, high fat diet, obesity etc and/or genetic predisposition to diabetes precipitate glucose intolerance. This is associated with islet dysfunction illustrated by increased proinsulin secretion seen in relatives of Type 2 diabetic subjects and in glucose-intolerant individuals. Without intervention, glucose intolerance progresses to diabetes with evidence of further aberrant islet function but not requiring amyloid formation or the associated beta-cell destruction as seen in animal models. Misprocessing of proinsulin and proIAPP as a result of a defect in beta-cell function leads to some fibril formation and some islets contain perivascular amyloid deposited adjacent to the basement membrane. Interaction between the abnormally functioning beta cell and small fibril accumulations and disrupted membrane signalling together induce further decline of islet function. Once fibrils are formed in the perivascular space, secreted IAPP will be rapidly refolded and create new fibrils. These fibrils or smaller toxic intermediates can induce apoptosis in adjacent cells and slowly (over decades) the islet cell population will be reduced. However, in diabetic individuals the time course of amyloid deposition and islet destruction is very variable suggesting that the causative factors for fibril accumulation could be intermittently apparent in islets of all diabetic subjects. The toxic effects of fibrils and induction of further fibril formation could be arrested by a protective coating of GAGs, ApoE and Amyloid P component on the perivascular fibrils. However, once islet cell loss associated with increased severity of amyloidosis becomes extensive, inadequate insulin production from the residual beta cells results in the requirement for insulin therapy
Fibril-induced cell death
Fibrils formed from synthetic IAPP and other amyloidogenic proteins rapidly show toxic properties to islet and other cells in vitro by an unknown mechanism [115, 116, 117]; insertion into the lipid bilayer and changes in cell membrane ion channel activity [118, 119] leading to apoptosis [12, 120], have been proposed. Both small oligomers of hIAPP and larger fibrils are thought to have cytotoxic properties [121, 122]. If non-fibrillar, small invisible hIAPP oligomers are the causative factor for death of beta cells in T2DM, it is surprising that there is not a severe reduction in islet mass in the absence of visible amyloidosis in man. In islets of Type 2 diabetic subjects cellular apoptosis has been identified [123] but other studies have shown that apoptotic cells are infrequent in adult islets [124]. The degree of toxicity induced by fibrils formed from secreted, biosynthetic human IAPP in hIAPP TM islets in short term (3 days) culture is minimal [105]; cells adjacent to fibrils show little evidence of cytotoxicity or apoptosis (Fig. 5) suggesting that the time course of synthetic fibril-induced toxicity is more rapid than that induced by biosynthetic hIAPP in vitro or in vivo. One suggestion is that the amyloid-associated proteins, SAP, ApoE, and glycosaminoglycans can act as protective factors in biological systems by reducing the impact of oligomers or fibrils on cell viability as well as reducing recognition for degradation or clearance of misfolded peptide [111]. However, rather than causing immediate cell death, small fibril accumulations could affect function; the beta-cell membranes adjacent to biosynthetic deposits in vivo (Fig. 2a) and in vitro (Fig. 5) are visibly disrupted which could interfere with cycling of membrane proteins and therefore stimulus–secretion coupling. Whether this association of fibrils with membranes is a cause or effect of cellular dysfunction in diabetes is not clear.
These data together suggest that amyloidosis and development of hyperglycaemia are similar, but not identical in man and animal models:
- (a):
-
In both man and in animal models, islet amyloid deposition commences as small perivascular deposits which progressively increase until many islets are affected. This perivascular stage is represented in many patients with long-term diabetes and in some non-diabetic subjects. However, in animals, this stage is usually associated with glucose intolerance and less frequently found in overt diabetes.
- (b):
-
More extensive amyloidosis develops when many islets are affected and is associated with cellular loss and the requirement for insulin therapy in diabetic humans but with onset of overt diabetes in hIAPP TM, monkeys and cats.
- (c):
-
Islet amyloid deposition, although apparently irreversible, is not related to the duration of hyperglycaemia in man.
- (d):
-
An environment of high glucose in diabetes does not, per se, result in fibril formation.
- (e):
-
Increased circulating hIAPP concentrations, whilst important, are not a causative factor for amyloidosis in either man or animal models.
Conclusion
The evidence suggests that islet amyloidosis is not a precipitating factor for hyperglycaemia in most patients with T2DM. However, in animal models of islet amyloid-related diabetes, glucose intolerance and hyperglycaemia can be directly related to the degree of amyloidosis. The factors that are responsible for oligomerisation of human IAPP in T2DM are poorly understood. Diabetes-related dysfunction of beta-cell metabolism or changes in the islet milieu are the most likely causes of IAPP fibril formation; amyloid deposition would then be a resultant pathological feature—a complication of diabetes (Fig. 7).
Since islet amyloidosis is the only visible factor that causes decline of islet function, how can its effects be reduced or prevented? The progressive nature of islet amyloidosis is consistent with the nucleation theory for all forms of amyloid deposition [125]. Once β-sheet oligomers, protofibrils or fibrils are formed, they can act as a nucleation point and promote more fibril formation from secreted peptide; conditions that result in increased secretion e.g. increased insulin resistance, sulphonylurea therapy in diabetes would then promote fibril formation. Islet amyloidosis was found to be more extensive in sulphonylurea-treated diabetic cats than those treated with insulin [126] confirming this hypothesis. However, the rate of deposition seems to be very different in human patients suggesting that the process is not be driven continuously from a nucleation point throughout the diabetic period.
The role of amyloid in the deterioration of islet function seems to be in conflict with the findings of the United Kingdom Prospective Diabetes Study, UKPDS, where treatment with diet, sulphonylurea or insulin was compared in 5000 newly-diagnosed patients [127]; there was no apparent difference in the rate of decline of islet function from the time of diagnosis in patient groups randomised to insulin or sulphonylurea treatment. However, since amyloid is not a precipitating factor for diabetes in man but more associated with the final demise of regulated insulin secretion (Fig. 7), it is unlikely that the decline in islet function over the first 10 years, especially in patients not requiring insulin therapy, would be caused by amyloid formation.
Assessment of efficacy of any therapeutic agent designed to reduce or prevent islet amyloid formation in man would require outcomes such as sustained islet function and decreased requirement for insulin therapy during the diabetic syndrome. This would require a large study of therapies such as that undertaken in the UKPDS. Even then, post-mortem evidence would be required to prove a reduction in amyloid formation since it cannot be assessed in vivo.
Some proposed targets for therapeutic intervention include:
- (i):
-
Prevent refolding of IAPP to form fibrils. So-called beta-sheet breaker peptides which bind specifically to amyloidogenic monomers to prevent refolding have been designed [128, 129]. These small compounds provide the best option for treatment of islet amyloidosis at present.
- (ii):
-
Removal of the deposits in vivo. Turnover and or removal of systemic amyloid deposits are thought to occur by macrophages or by increased solubilisation of the fibrils. If phagocytosis of amyloid deposits could be enhanced by removal of factors that prevent recognition by macrophages, islet amyloidosis could be reduced by a normal immune response to aberrant protein aggregation.
The factors that destabilise the conformation of this little peptide and cause such destructive changes in islets in the course of the disease are dramatic and seem to be specific for T2DM. This suggests that islet amyloidosis should be added to the long list of complications of T2DM that increase both morbidity and mortality. More than a century after the identification of islet hyaline deposition in diabetes [4] the casual mechanisms for this islet pathology are still unclear. With the inexorable increase in incidence of T2DM, it is essential that the new technologies of proteomics, metabolomics and biophysics provide a detailed understanding of normal islet biochemistry and predict changes in function that result in hyperglycaemia. Will it take another century before we can solve the scientific enigma of islet amyloid deposition?
Sources
This review is based upon studies reported in the literature over the last 20 years and those made in Oxford by the authors, their colleagues, students and by international collaborators over a similar period. Some unpublished data included in D Phil theses are available through the Bodelian Library, University of Oxford.
Abbreviations
- IAPP:
-
islet amyloid polypeptide
- hIAPP:
-
human islet amyloid polypeptide
- T2DM:
-
Type 2 diabetes
- TM:
-
transgenic mice expressing the human IAPP gene
- UKPDS:
-
United Kingdom Prospective Diabetes Study
- GAGs:
-
glycosamino glycans
References
Schneider HM, Storkel S, Will W (1980) Das Amyloid der Langerhansschen Inseln und seine Beziehung zum Diabetes mellitus. Dtsch Med Wochenschr 105:1143–1147
Röcken C, Linke RP, Saeger W (1992) Immunohistology of islet amyloid polypeptide in diabetes mellitus: semi-quantitative studies in a post-mortem series. Virchows Arch A Pathol Anat Histopathol 421:339–344
Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC (1988) Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res Clin Prac 9:151–159
Opie E (1901) The relation of diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands of Langerhans. J Exp Med 5:527–540
Cecil RL (1909) A study of the pathological anatomy of the pancreas in ninety cases of diabetes mellitus. J Exp Med 11:266–290
Ehrlich J, Ratner M (1961) Amyloidosis of the islets of Langerhans. A restudy of islet hyaline in diabetic and non-diabetic individuals. Am J Pathol 38:49–59
Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ (1999) Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19:8876–8884
Heitner J, Dickson D (1997) Diabetics do not have increased Alzheimer-type pathology compared with age-matched control subjects. A retrospective postmortem immunocytochemical and histofluorescent study. Neurology 49:1306–1311
Bruce DG, Harrington N, Davis WA, Davis TM (2001) Dementia and its associations in type 2 diabetes mellitus: the Fremantle Diabetes Study. Diabetes Res Clin Pract 53:165–172
Kahn SE, Andrikopoulos S, Verchere CB (1999) Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48:241–253
Westermark P (1994) Amyloid and polypeptide hormones: what is their interrelationship? Int J Exp Clin Invest 1:47–60
Lorenzo A, Razzaboni B, Weir G, Yankner B (1994) Pancreatic islet cell toxicity of amylin associated with Type-2 diabetes mellitus. Nature 368:756–760
Janciauskiene S, Ahrén B (1998) Different sensitivity to the cytotoxic action of IAPP fibrils in two insulin-producing cell lines, HIT-T15 and RINm5F cells. Biochem Biophys Res Commun 251:888–893
Sanke T, Hanabusa T, Nakano Y, Oki C, Okai K, Nishimura S, Kondo M, Nanjo K (1991) Plasma islet amyloid polypeptide (amylin) levels and their responses to oral glucose in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 34:129–132
Butler PC, Chou J, Carter WB, Wang YN, Bu BH, Chang D, Chang JK, Rizza RA (1990) Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes 39:752–756
Kahn SE, Verchere CB, Andrikopoulos S, Asberry PJ, Leonetti DL, Wahl PW, Boyko EJ, Schwartz RS, Newell-Morris L, Fujimoto WY (1998) Reduced amylin release is a characteristic of impaired glucose tolerance and type 2 diabetes in Japanese Americans. Diabetes 47:640–645
Sanke T, Bell GI, Sample C, Rubenstein AH, Steiner DF (1988) An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J Biol Chem 263:17243–17246
Badman MK, Shennan KI, Jermany JL, Docherty K, Clark A (1996) Processing of pro-islet amyloid polypeptide (proIAPP) by the prohormone convertase PC2. FEBS Lett 378:227–231
Higham. CE, Hull RL, Lawrie L, Shennan KIJ, Morris JF, Birch NP, Docherty K, Clark A (2000) Processing of synthetic pro-islet amyloid polypeptide (proIAPP) ‘amylin’ by recombinant prohormone convertase enzymes, PC2 and PC3, in vitro. Eur J Biochem 267:4998–5004
Hutton JC (1994) Insulin secretory granule biogenesis and the proinsulin-processing endopeptidases. Diabetologia 37 [Suppl 2]:S48–56
Kautzky-Willer A, Thomaseth K, Pacini G, Clodi M, Ludvik B, Streli C, Waldhausl W, Prager R (1994) Role of islet amyloid polypeptide secretion in insulin-resistant humans. Diabetologia 37:188–194
Jaikaran ETAS, Clark A (2001) Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim Biophys Acta 1537:179–203
Koning EJ de, Bodkin NL, Hansen BC, Clark A (1993) Diabetes mellitus in Macaca mulatta monkeys is characterised by islet amyloidosis and reduction in beta-cell population. Diabetologia 36:378–384
Howard C (1978) Insular amyloidosis and diabetes mellitus in Macaca nigra. Diabetes 27:357–364
O’Brien T, Hayden D, Johnson K, Fletcher T (1986) Immuno-histochemical morphometry of pancreatic endocrine cells in diabetic normoglycaemic glucose-intolerant and normal cats. J Comp Pathol 96:357–369
Betsholtz C, Christmansson L, Engström U, Rorsman F, Svensson V, Johnson KH, Westermark P (1989) Sequence divergence in a specific region of islet amyloid polypeptide (IAPP) explains differences in islet amyloid formation between species. FEBS Lett 251:261–264
Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C (1990) Islet amyloid polypeptide—pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA 87:5036–5040
Moriarty DF, Raleigh DP (1999) Effects of sequential proline substitutions on amyloid formation by human amylin (20–29). Biochemistry 38:1811–1818
Green J, Goldsbury C, Mini T, Sunderji S, Frey P, Kistler J, Cooper G, Aebi U (2003) Full-length rat amylin forms fibrils following substitution of single residues from human amylin. J Mol Biol 326:1147–1156
Roberts A, Leighton B, Todd J, Cockburn D, Schofield P, Sutton R, Holt S, Boyd Y, Day A, Foot E, et al. (1989) Molecular and functional characterization of amylin, a peptide associated with type 2 diabetes mellitus. Proc Natl Acad Sci USA 86:9662–9666
Bushfield M, Savage A, Morris NJ, Houslay MD (1993) A mnemonical or negative-co-operativity model for the activation of adenylate cyclase by a common G-protein-coupled calcitonin-gene-related neuropeptide (CGRP)/amylin receptor. Biochem J 293:229–236
Bhogal R, Smith DM, Bloom SR (1992) Investigation and characterisation of binding sites for islet amyloid polypeptide in rat membranes. Endocrinology 130:906–913
Zumpe ET, Tilakaratne N, Fraser NJ, Christopoulos G, Foord SM, Sexton PM (2000) Multiple ramp domains are required for generation of amylin receptor phenotype from the calcitonin receptor gene product. Biochem Biophys Res Commun 267:368–372
Clark A, Cooper GJ, Lewis CE, Morris JF, Willis AC, Reid KB, Turner RC (1987) Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet 2:231–234
Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KH (1987) Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci USA 84:3881–3885
Gebre-Medhin S, Olofsson C, Mulder H (2000) Islet amyloid polypeptide in the islets of Langerhans: friend or foe? Diabetologia 43:687–695
Gebre-Medhin S, Mulder H, Pekny M, Westermark G, Tornell J, Westermark P, Sundler F, Ahrén B, Betsholtz C (1998) Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem Biophys Res Commun 250:271–277
Ahrén B, Oösterwijk C, Lips CJ, Höppener JW (1998) Transgenic overexpression of human islet amyloid polypeptide inhibits insulin secretion and glucose elimination after gastric glucose gavage in mice. Diabetologia 41:1374–1380
Maloy AL, Longnecker DS, Greenberg ER (1981) The relation of islet amyloid to the clinical type of diabetes. Hum Pathol 12:917–922
Clark A, Holman R, Matthews D, Hockaday T, Turner R (1984) Non-uniform distribution of islet amyloid in the pancreas of ‘maturity-onset’ diabetic patients. Diabetologia 27:527–528
Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, Lovat LB, Bartfai T, Alanine A, Hertel C, Hoffmann T, Jakob-Roetne R, Norcross RD, Kemp JA, Yamamura K, Suzuki M, Taylor GW, Murray S, Thompson D, Purvis A, Kolstoe S, Wood SP, Hawkins PN (2002) Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 417:254–259
Chargé SB, Esiri MM, Bethune CA, Hansen BC, Clark A (1996) Apolipoprotein E is associated with islet amyloid and other amyloidoses: implications for Alzheimer’s disease. J Pathol 179:443–447
Snow AD, Willmer J, Kisilevsky R (1987) Sulfated glycosaminoglycans: a common constituent of all amyloids? Lab Invest 56:120–123
Wisniewski T, Frangione B (1992) Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 135:235–238
Rochet JC, Lansbury PT Jr (2000) Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol 10:60–68
Higham CE, Jaikaran ETAS, Fraser PE, Gross M, Clark A (2000) Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Letts 470:55–60
Couce M, Kane LA, O’Brien TD, Charlesworth J, Soeller W, McNeish J, Kreutter D, Roche P, Butler PC (1996) Treatment with growth hormone and dexamethasone in mice transgenic for human islet amyloid polypeptide causes islet amyloidosis and beta-cell dysfunction. Diabetes 45:1094–1101
Westermark P, Eizirik DL, Pipeleers DG, Hellerström C, Andersson A (1995) Rapid deposition of amyloid in human islets transplanted into nude mice. Diabetologia 38:543–549
Koning EJ de, Morris ER, Hofhuis FM, Posthuma G, Hoppener JW, Morris JF, Capel PJ, Clark A, Verbeek JS (1994) Intra- and extracellular amyloid fibrils are formed in cultured pancreatic islets of transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 91:8467–8471
Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kapurniotu A (1999) Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J Mol Biol 287:781–796
Goldsbury C, Kistler J, Aebi U, Arvinte T, Cooper GJ (1999) Watching amyloid fibrils grow by time-lapse atomic force microscopy. J Mol Biol 285:33–39
Nishi M, Sanke T, Nagamatsu S, Bell GI, Steiner DF (1990) Islet amyloid polypeptide. A new b-cell secretory product related to islet amyloid deposits. J Biol Chem 265:4173–4176
Hutton JC (1984) Secretory granules. Experientia 40:1091–1098
Janciauskiene S, Eriksson S, Carlemalm E, Ahren B (1997) B cell granule peptides affect human islet amyloid polypeptide (IAPP) fibril formation in vitro. Biochem Biophys Res Commun 236:580–585
Westermark P, Li ZC, Westermark GT, Leckstrom A, Steiner DF (1996) Effects of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett 379:203–206
Jaikaran ETAS, Nilsson MR, Clark A (2003) Pancreatic b-cell granule peptides form heteromolecular complexes which inhibit islet amyloid polypeptide fibril formation. Biochem J Oct 20 (epublication)
Larson JL, Ko E, Miranker AD (2000) Direct measurement of islet amyloid polypeptide fibrillogenesis by mass spectrometry. Protein Sci 9:427–431
Jaikaran ETAS, Higham CE, Serpell LC, Zurdo J, Gross M, Clark A, Fraser PE (2001) Identification of a novel human islet amyloid polypeptide beta-sheet domain and factors influencing fibrillogenesis. J Mol Biol 308:515–525
Serpell LC, Sunde M, Blake CCF (1997) The molecular basis of amyloidosis. Cell Mol Life Sci 53:871–887
Nilsson MR, Driscoll M, Raleigh DP (2002) Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: implications for the study of amyloid formation. Protein Sci 11:342–349
Nishi M, Bell GI, Steiner DF (1990) Islet amyloid polypeptide (amylin): no evidence of an abnormal precursor sequence in 25 type 2 (non-insulin-dependent) diabetic patients. Diabetologia 33:628–630
Cook JT, Patel PP, Clark A, Hoppener JW, Lips CJ, Mosselman S, O’Rahilly S, Page RC, Wainscoat JS, Turner RC (1991) Non-linkage of the islet amyloid polypeptide gene with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 34:103–108
Seino S (2001) S20G mutation of the amylin gene is associated with Type II diabetes in Japanese. study group of comprehensive analysis of genetic factors in diabetes mellitus. Diabetologia 44:906–909
Sakagashira S, Sanke T, Hanabusa T, Shimomura H, Ohagi S, Kumagaye KY, Nakajima K, Nanjo K (1996) Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 45:1279–1281
Lee SC, Hashim Y, Li JKY, Ko GTC, Critchley JAJH, Cockram CS, Chan JCN (2001) The islet amyloid polypeptide (amylin) gene S20G mutation in Chinese subjects: evidence for associations with type 2 diabetes and cholesterol levels. Clin Endocrinol 54:541–546
Birch CL, Fagan LJ, Armstrong MJ, Turnbull DM, Walker M (1997) The S20G islet-associated polypeptide gene mutation in familial NIDDM (Letter). Diabetologia 40:1113
Yamada K, Yuan X, Ishiyama S, Nonaka K (1998) Glucose tolerance in Japanese subjects with S20G mutation of the amylin gene. Diabetologia 41:125
Westermark P, Ma Z, Gustavsson A,Westermark GT, Engström U, Sakagashira S, Sanke T, Nanjo K (1998) Enhanced in vitro production of amyloid-like fibrils from mutant (S20G) islet amyloid polypeptide. In: Kyle RA, Gertz MA (eds) Amyloid and amyloidosis. Proceedings of the VIIIth International Symposium on Amyloidosis. Parthenon, New York, pp 551–553
Sakagashira S, Hiddinga HJ, Tateishi K, Sanke T, Hanabusa T, Nanjo K, Eberhardt NL (2000) S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. Am J Pathol 157:2101–2109
Novials A, Rojas I, Casamitjana R, Usac EF, Gomis R (2001) A novel mutation in islet amyloid polypeptide (IAPP) gene promoter is associated with Type II diabetes mellitus. Diabetologia 44:1064–1065
Clark A, Matthews DR, Naylor BA, Wells CA, Hosker JP, Turner RC (1987) Pancreatic islet amyloid and elevated proinsulin secretion in familial maturity-onset diabetes. Diabetes Res Clin Prac 4:51–55
Porte D, Kahn SE (1989) Hyperproinsulinaemia and amyloid in NIDDM: clues to etiology of islet beta-cell dysfunction. Diabetes 38:1333–1336
Halban PA, Kahn SE (1997) Release of incompletely processed proinsulin is the cause of the disproportionate proinsulineamia of NIDDM. Diabetes 46:1725–1732
Hou X, Ling Z, Quartier E, Foriers A, Schuit F, Pipeleers D, Van Schravendijk C (1999) Prolonged exposure of pancreatic beta cells to raised glucose concentrations results in increased cellular content of islet amyloid polypeptide precursors. Diabetologia 42:188–194
Westermark GT, Steiner DF, Gebre-Medhin S, Engström U, Westermark P (2000) Pro islet amyloid polypeptide (proIAPP) immunoreactivity in the islets of Langerhans. Uppsala J Med Sci 105:97–106
Wang J, Xu J, Finnerty J, Furuta M, Steiner DF, Verchere CB (2001) The prohormone convertase enzyme 2 (PC2) is essential for processing pro-islet amyloid polypeptide at the NH2-terminal cleavage site. Diabetes 50:534–539
Krampert M, Bernhagen J, Schmucker J, Horn A, Schmauder A, Brunner H, Voelter W, Kapurniotu A (2000) Amyloidogenicity of recombinant human pro-islet amyloid polypeptide (ProIAPP). Chem Biol 7:855–871
Park K, Verchere CB (2000) Identification of a heparin-binding domain in the N-terminal cleavage site of pro-islet amyloid polypeptide: implications for islet amyloid formation. J Biol Chem 276:16611–16616
Potter-Perigo S, Hull RL, Tsoi C, Braun KR, Andrikopoulos S, Teague J, Verchere CB, Kahn SE Wight TN (2003) Proteoglycans synthesized and secreted by pancreatic islet beta-cells bind amylin. Arch Biochem Biophys 413:182–190
Furth AJ (1997) Glycated proteins in diabetes. Br J Biomed Sci 54:192–200
McKillop AM, Mooney MH, Harriott P, Flatt PR, O’Harte FP (2001) Evaluation of glycated insulin in diabetic animals using immunocytochemistry and radioimmunoassay. Biochem Biophys Res Commun 286:524–528
Kapourniotu A, Bernhagen J, Greenfield N, Al-Abed Y, Teichberg S, Frank RW, Voelter W, Bucala R (1998) Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. Eur J Biochem 251:208–216
Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, Kindy M (2000) Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med 6:643–651
Enoki S, Mitsukawa T, Takemura J, Nakazato M, Aburaya J, Tosgiori H, Matsukara S (1992) Plasma islet amyloid polypeptide levels in obesity, impaired glucose tolerance and non-insulin dependent diabetes mellitus. Diabetes Res Clin Pract 15:97–102
German MS, Moss LG, Wang J, Rutter WJ (1992) The insulin and islet amyloid polypeptide genes contain similar cell-specific promoter elements that bind identical beta-cell nuclear complexes. Mol Cell Biol 12:1777–1788
Hartter E, Svoboda T, Ludvik B, Schuller M, Lell B, Kuenburg E, Brunnbauer M, Woloszczuk W, Prager R (1991) Basal and stimulated plasma levels of pancreatic amylin indicate its co-secretion with insulin in humans. Diabetologia 34:52–54
Jorgensen JO, Rosenfalck AM, Fisker S, Nyholm B, Fineman MS, Schmitz O, Madsbad S, Holst JJ, Christiansen JS (2000) Circulating levels of incretin hormones and amylin in the fasting state and after oral glucose in GH-deficient patients before and after GH replacement: a placebo-controlled study. Eur J Endocrinol 143:593–599
Mulder H, Ahrén B, Stridsberg M, Sundler F (1995) Non-parallelism of islet amyloid polypeptide (amylin) and insulin gene expression in rat islets following dexamethasone treatment. Diabetologia 38:395–402
O’Brien TD, Westermark P, Johnson KH (1991) Islet amyloid polypeptide and insulin secretion from isolated perfused pancreas of fed, fasted, glucose-treated, and dexamethasone-treated rats. Diabetes 40:1701–1706
Ludvik B, Clodi M, Kautzky-Willer A, Capek M, Hartter E, Pacini G, Prager R (1993) Effect of dexamethasone on insulin sensitivity, islet amyloid polypeptide and insulin secretion in humans. Diabetologia 36:84–87
Fox N, Schrementi J, Nishi M, Ohagi S, Chan SJ, Heisserman JA, Westermark GT, Leckstrom A, Westermark P, Steiner DF (1993) Human islet amyloid polypeptide transgenic mice as a model of non-insulin-dependent diabetes mellitus (NIDDM). FEBS Lett 323:40–44
D’Alessio DA, Verchere CB, Kahn SE, Hoagland V, Baskin DG, Palmiter RD, Ensinck JW (1994) Pancreatic expression and secretion of human islet amyloid polypeptide in a transgenic mouse. Diabetes 43:1457–1461
Höppener JW, Verbeek JS, Koning EJ de, Oösterwijk C, Hulst KL van, Visser-Vernooy HJ, Hofhuis FM, Gaalen S van, Berends MJ, Hackeng WH, et al. (1993) Chronic overproduction of islet amyloid polypeptide/amylin in transgenic mice: lysosomal localization of human islet amyloid polypeptide and lack of marked hyperglycaemia or hyperinsulinaemia. Diabetologia 36:1258–1265
Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, Butler PC (1996) Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 93:7283–7288
Höppener JWM, Oösterwijk C, Nieuwenhuis MG, Posthuma G, Thijssen JHH, Vroom TM, Ahrén B, Lips CJM (1999) Extensive islet amyloid formation is induced by development of type II diabetes mellitus and contributes to its progression: pathogenesis of diabetes in a mouse model. Diabetologia 42:427–434
Soeller WC, Janson J, Hart SE, Parker JC, Carty MD, Stevenson RW, Kreutter DK, Butler PC (1998) Islet amyloid-associated diabetes in obese A(vy)/a mice expressing human islet amyloid polypeptide. Diabetes 47:743–750
Verchere CB, D’Alessio DA, Palmiter RD, Weir GC, Bonner Weir S, Baskin DG, Kahn SE (1996) Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc Natl Acad Sci USA 93:3492–3496
Geisler JG, Zawalich W, Zawalich K, Lakey JR, Stukenbrok H, Milici AJ, Soeller WC (2002) Estrogen can prevent or reverse obesity and diabetes in mice expressing human islet amyloid polypeptide. Diabetes 51:2158–2169
Hull RL, Verchere CB, Andrikopoulos S, Wang F, Vidal J, Kahn SE (2001) Oophorectomy promotes islet amyloid formation in human islet amyloid polypeptide transgenic mice islet. Diabetes 50 [Suppl 1]:S184–185
Westermark GT, Gebre-Medhin S, Steiner DF, Westermark P (2000) Islet amyloid development in a mouse strain lacking endogenous islet amyloid polypeptide (IAPP) but expressing human IAPP. Mol Med 6:998–1007
Ma Z, Westermark GT (2002) Effects of free fatty acid on polymerization of islet amyloid polypeptide (IAPP) in vitro and on amyloid fibril formation in cultivated isolated islets of transgenic mice overexpressing human IAPP. Mol Med 8:863–868
Hull RL, Andrikopoulos S, Verchere CB, Vidal J, Wang F, Cnop M, Prigeon RL, Kahn SE (2003) Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes 52:372–379
Vidal J, Verchere CB, Andrikopoulos S, Wang F, Hull RL, Cnop M, Olin KL, LeBoeuf RC, O’Brien KD, Chait A, Kahn SE (2003) The effect of apolipoprotein E deficiency on islet amyloid deposition in human islet amyloid polypeptide transgenic mice. Diabetologia 46:71–79
Clark A, Saad MF, Nezzer T, Uren C, Knowler WC, Bennett PH, Turner RC (1990) Islet amyloid polypeptide in diabetic and non-diabetic Pima Indians. Diabetologia 33:285–289
MacArthur DLA, Koning EJP de, Verbeek JS, Morris JE, Clark A (1999) Amyloid fibril formation is progressive and correlates with beta-cell secretion in transgenic mouse isolated islets. Diabetologia 42:1219–1227
Carlsson P-O, Jansson L, Ostenson C-G, Kallskog O (1997) Islet capillary blood pressure increase mediated by hyperglycaemia in NIDDM GK rats. Diabetes 46:947–952
Schnell AH, Swenne I, Borg LAH (1988) Lysosomes and pancreatic islet function: a quantitative estimation of crinophagy in the mouse pnacreatic beta cell. Cell Tissue Res. 252:9–15
Clark A, Edwards CA, Ostle LR, Sutton R, Rothbard JB, Morris JF, Turner RC (1989) Localisation of islet amyloid peptide in lipofuscin bodies and secretory granules of human beta-cells and in islets of type-2 diabetic subjects. Cell Tissue Res 257:179–185
Koning EJ de, Höppener JW, Verbeek JS, Oösterwijk C, Hulst KL van, Baker CA, Lips CJ, Morris JF, Clark A (1994) Human islet amyloid polypeptide accumulates at similar sites in islets of transgenic mice and humans. Diabetes 43:640–644
Pepys MB (2001) Pathogenesis, diagnosis and treatment of systemic amyloidosis. Philos Trans R Soc Lond B Biol Sci 356:203–210
Botto M, Hawkins PN, Bickerstaff MC, Herbert J, Bygrave AE, McBride A, Hutchinson WL, Tennent GA, Walport MJ, Pepys MB (1997) Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat Med 3:855–859
Badman MK, Pryce RA, Charge SB, Morris JF, Clark A (1998) Fibrillar islet amyloid polypeptide (amylin) is internalised by macrophages but resists proteolytic degradation. Cell Tissue Res 291:285–294
Koning EJ de, Brand JJ van den, Mott VL, Chargé SB, Hansen BC, Bodkin NL, Morris JF, Clark A (1998) Macrophages and pancreatic islet amyloidosis. Amyloid 5:247–254
Wang F, Hull RL, Vidal J, Cnop M, Kahn SE (2001) Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes 50:2514–2520
Caughey B, Lansbury PT Jr (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298
Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC (1999) The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 48:491–498
Scrocchi LA, Ha K, Chen Y, Wu L, Wang F, Fraser PE (2003) Identification of minimal peptide sequences in the (8–20) domain of human islet amyloid polypeptide involved in fibrillogenesis. J Struct Biol 141:218–227
Mirzabekov T, Lin M, Kagan B (1996) Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem 271:1988–1992
Wagoner PK, Chen C, Worley JF, Dukes ID, Oxford GS (1993) Amylin modulates beta-cell glucose sensing via effects on stimulus-secretion coupling. Proc Natl Acad Sci USA 90:9145–9149
Saafi EL, Konarkowska B, Zhang S, Kistler J, Cooper GJ (2001) Ultrastructural evidence that apoptosis is the mechanism by which human amylin evokes death in RINm5F pancreatic islet beta-cells. Cell Biol Int 25:339–350
Harroun TA, Bradshaw JP, Ashley RH (2001) Inhibitors can arrest the membrane activity of human islet amyloid polypeptide independently of amyloid formation. FEBS Lett 507:200–204
Anguiano M, Nowak RJ, Lansbury PT Jr (2002) Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 41:11338–11343
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B (2000) Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49:1325–1333
Jarrett JT, Lansbury PT Jr (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73:1055–1058
Hoenig M, Hall G, Ferguson F, Jordan K, Henderson M, Johnson K, O’Brien T (2000) A feline model of experimentally induced amyloidosis. Am J Pathol 157:2143–2150
UKPDS (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B (1998) Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat Med 4:822–826
Scrocchi LA, Chen Y, Waschuk S, Wang F, Cheung S, Darabie AA, McLaurin J, Fraser PE (2002) Design of peptide-based inhibitors of human islet amyloid polypeptide fibrillogenesis. J Mol Biol 318:697–706
Acknowledgements
We are grateful to Professors J. Morris (Oxford), C. Dobson (Cambridge) and P. Fraser (Toronto) for their continued support of our projects. We thank B. Hansen (Baltimore, USA) and the Oxford students C. Higham, E. Jaikaran, R. Bhogal, E. de Koning, J. Moffitt and M.K. Badman for contributing unpublished data. We are grateful to The Wellcome Trust (AC & MRN) for financial support. The electron microscope in the Department of Human Anatomy and Genetics was purchased by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clark, A., Nilsson, M.R. Islet amyloid: a complication of islet dysfunction or an aetiological factor in Type 2 diabetes?. Diabetologia 47, 157–169 (2004). https://doi.org/10.1007/s00125-003-1304-4
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1304-4