
Abstract
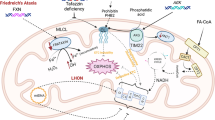
Mitochondria are the major sites of energy production in the cell as they harbor the process of oxidative phosphorylation (OXPHOS). OXPHOS is performed by proteins at the mitochondrial respiratory chain, comprising complexes I–IV and adenosine triphosphate (ATP) synthase (complex V). As the heart is an energy-dependent tissue, mitochondria constitute 20–40% of the cellular volume of cardiomyocytes. The mitochondrial energy production is under the genetic control of both nuclear and mitochondrial genes. Mutations within these genes may cause defects in oxidative phosphorylation and have severe consequences for those organs which are heavily dependent on energy production like the heart, the brain, and skeletal muscle. Because myopathy is often one of the main presenting symptoms, patients with mitochondrial diseases tend to be seen primarily by neurologists and pediatricians. However, the importance of mitochondrial disease in cardiology is being more and more recognized, not only because cardiomyopathy may be the only manifestation of mitochondrial disease (Fig. 7.1).
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am Heart J. 2000;139:S70-S85.
Ashizawa T. What is Kearns-Sayre syndrome after all? Arch Neurol. 2001;58:1053-1054.
Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925-931.
Garcia J-M, Goldenthal MJ. Understanding the impact of mitochondrial defects in cardiovascular disease: a review. J Cardiac Failure. 2002;8:347-361.
Holmgren D, Wahlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M. Cardiomyopathy in children with mitochondrial disease. Eur Heart J. 2003;24:280-288.
Ware SM, El-Hassan N, Kahler SG, et al. Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes. J Med Genet. 2009;46:308-314.
Yaplito-Lee J, Weintraub R, Jamsen K, et al. Cardiac manifestations in oxidative phosphorylation disorders of childhood. J Pediatr. 2007;150:407-411.
Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984;16:481-488.
Goto Y, Nonaka I, Horai S. A mutation in the tRNA (Leu) (UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651-653.
Sproule D, Kaufman P. Mitochondrial encephalopathy, lactic acidosis and strokelike episodes. Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann NY Acad Sci. 2008;1142:133-158.
Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) current concepts. J Child Neurol. 1994;9:4-13.
Vydt TCG, de Coo RFM, Soliman OII, ten Cate FJ, van Geuns RJM, et al. Cardiac involvement in adults with m.3242A>G MELAS gene mutation. Am J Card. 2007;99:264-269.
Wortmann SB, Rodenburg RJ, Backx AP, Schmitt E, Smeitink JAM, Morava E. Early cardiac involvement in children carrying the A3243G mtDNA mutation. Acta Paediatrica. 2007;96:450-451.
Okajima Y, Tanabe Y, Takayanagi M, Aotsuka H. A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS). Heart. 1998;80:292-295.
Sproule DM, Kaufmann P, Engelstad K, et al. Wolff-Parkinson-White syndrome in patients with MELAS. Arch Neurol. 2007;64:1625-1627.
Shanske S, Pancrudo J, Kaufmann P, et al. Varying loads of mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet. 2004;130A:134-137.
Bhati RS, Sheridan BC, Mill MR, Selzman CH. Heart transplantation for progressive cardiomyopathy as a manifestation of MELAS syndrome. J Heart Lung Transplant. 2005;24:2286-2289.
Muravchick S. Clinical implications of mitochondrial disease. Adv Drug Deliv Rev. 2008;60:1553-1560.
Finsterer J. Mitochondriopathies. Eur J Neurol. 2004;11:163-186.
Roberts NK, Perloff JK, Kark RAP. Cardiac conduction in the Kearns-Sayre Syndrome (a neuromuscular disorder associated with progressive external ophthalmoplegia and pigmentary retinopathy). Am J Cardiol. 1979;44:1396-1400.
Anan R, Nakagawa M, Miyata M, Higuchi I, Nakao S, et al. Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation. 1995;91:955-961.
Welzing L, von Kleist-Retzow JC, Kribs A, et al. Rapid development of life-threatening complete atrioventricular block in Kearns-Sayre syndrome. Eur J Pediatr. 2009;168:757-759.
Chawla S, Coku J, Forbes T, Kannan S. Kearns-Sayre syndrome presenting as complete heart block. Pediatr Cardiol. 2008;29:659-662.
Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device based therapy of cardiac rhythm abnormalities. J Am Coll Cardiol. 2008;51:e1-e62.
Charles R, Holt S, Kay JM, et al. Myocardial ultrastucture and the development of atrioventricular block in Kearns-Sayre syndrome. Circulation. 1981;63:214-219.
Channer KS, Channer JL, Campbell MJ, Russel RJ. Cardiomyopaty in the Kearns-Sayre syndrome. Br Heart J. 1988;59:486-490.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Verlag London Limited
About this chapter
Cite this chapter
de Jonge, N., Kirkels, J.H. (2011). Mitochondrial Cardiomyopathy. In: Baars, H., Doevendans, P., van der Smagt, J. (eds) Clinical Cardiogenetics. Springer, London. https://doi.org/10.1007/978-1-84996-471-5_7
Download citation
DOI: https://doi.org/10.1007/978-1-84996-471-5_7
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-84996-470-8
Online ISBN: 978-1-84996-471-5
eBook Packages: MedicineMedicine (R0)